Single-cell Fixed RNA sequencing from Formalin-Fixed, Paraffin-Embedded (FFPE)-Lung Tissue by Chromium Fixed RNA Profiling for Multiplexed Samples
Mauricio Rojas, Lorena Rosas, Ana L. Mora, Natalia-Del Pilar Vanegas, Jose A. Ovando-Ricardez
Abstract
Fixed RNA Profiling provides an approach to FFPE-lung tissue samples that were previously inaccessible. Fixation at the point of sample collection preserves fragile biology to provide high-quality data on gene expression of multiple cell types in the lung, generating critical and novel insights. This protocol describes the new platform for single cell RNA-seq from Fixed RNA, which uses fixed cells isolated from paraffin-embedded tissue and multiplexed samples to analyze multiple samples in a single run to generate DNA libraries for sequencing.
Steps
Isolation of Cells from FFPE Tissue Sections for Chromium Fixed RNA Profiling According to Demonstrated Protocol CG000632 by 10x genomics.
Protocol
Prepare two 50 μm sections from the rehydrated FFPE tissue block and transfer them into a gentleMACSTM C Tube while keeping the scrolls intact.
Add 3 ml xylene to the gentleMACSTM C Tube and incubate for 10 min at RT.
Remove the liquid from the tube without breaking the scrolls.
Repeat steps b-c twice.
Add 3 ml 100% ethanol to the gentleMACSTM C Tube and incubate for 30 sec at RT.
Remove the liquid from the tube without breaking the scrolls.
Add 1 ml 100% ethanol and incubate for 30 sec at RT.
Remove the liquid from the tube without breaking the scrolls.
Add 1 ml 70% ethanol and incubate for 30 sec at RT.
Remove the liquid from the tube without breaking the scrolls.
Add 1 ml 50% ethanol and incubate for 30 sec at RT.
Remove the liquid from the tube without breaking the scrolls.
Add 1 ml nuclease-free water to the tube and incubate for 30 sec at RT.
Remove the liquid from the tube without breaking the scrolls.
Add 1 ml PBS and maintain on ice.
Remove the PBS from the gentleMACSTM C Tube without breaking the scrolls.
Add 2 ml Dissociation Enzyme Mix to the gentleMACSTM C Tube and close securely.
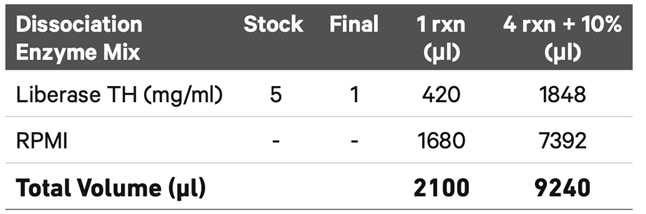
Place the tube on the gentleMACSTM Octo Dissociator, apply Heating units, and run the gentleMACSTM Program 37C_FFPE_1. Run time ~48 min.

At the end of the run, detach the tube from the gentleMACSTM Octo Dissociator.
Centrifuge at ~300 rcf for 1 min and resuspend the cell pellet in the supernatant.
Pass the suspension through a Pre-Separation Filter (30 μm) placed on a 15-ml tube on ice.
Rinse the original gentleMACSTM tube with 2 ml chilled PBS and use that rinse for an additional wash of the 30 μm filter to minimize cell loss. Collect the filtrate in the same tube.
Centrifuge the cell suspension at 850 rcf at 4°C for 5 min.
Remove the supernatant without disturbing the pellet.
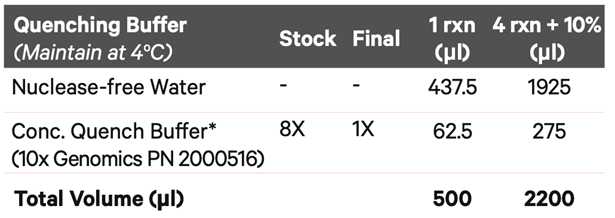
Resuspend the pellet in 0.5 ml chilled Quenching Buffer, pipette mix 5x, and maintain on ice.
Determine the cell concentration of the fixed sample using an Automated Cell Counter.
Proceed immediately to appropriate Chromium Fixed RNA for Multiplexed Samples using 16 Probe Barcodes – Probe Hybridization step 1.1 ( see User Guide CG000527 Rev D by 10x Genomics )
Data analyis
The FASTQ files from sequencing will be processed using Cell Ranger 7.1.0 from 10x Genomics. The Cell Ranger count feature will be employed for read alignment and count calculation, utilizing the human reference genome (GRCh38, GENCODE v32/Ensembl 98).
Treat each sample as an individual batch. Conduct all post-alignment analyses within an R 4.2.3 environment and Bioconductor 3.17. Specifically, generate R objects for each different slide using Seurat 4.3.0.
Calculate the percentage of genes related to mitochondria, ribosome, and hemoglobin. and remove cells with outliers.
Normalize individual samples using the negative binomial model SCTransform, then merge the Seurat objects.
Employ the anchor identification method to integration, considering 3000 genes as highly variable. Determine anchors for each sample using the FindIntegrationAnchors() function, and then proceed with integration using IntegrateData().
Identify the principal components (PCs) of our integrated object using the RunPCA() function. Define the number of dimensions using an Elbow plot with the most significant PCs for further analysis.
Reduce the dimensions of the data using the most significant PCs with the UMAP method. Afterward, find the nearest neighbors and perform clustering.
The clusters will be annotated according to the markers suggested by the Human Lung Cell Atlas and LungMAP1,2.
