RNA extraction with spin columns from yeast cells grown on 12-column deep well plates
Jamie Auxillos, Rosey Bayne, Edward Wallace
Abstract
This protocol is for yeast cell growth, lysis, and RNA extraction using silica spin columns. We use this to prepare RNA for RT-qPCR analysis from many strains at once. We have particularly used it on S288C-background Saccharomyces cerevisiae , containing a plasmid encoding a fluorescent reporter and a URA3 selection cassette, grown in SC-URA media. Then we use RT-qPCR to measure reporter gene expression compared to reference genes. In principle the protocol should work for similar strains/species and growth media, as long as the cell shape and cell walls are similar enough that the lysis method works.
Yeast strains are grown in parallel, in a 12 column deep well plate, on a shaking incubator allowing for easier strain handling. Yeast cells then are lysed by bead bashing with zirconia beads using the Precellys Evolution homogeniser. RNA is subsequently purified using a silica column. The RNA extraction method was a modification of the Quick-DNA/RNA miniprep kit from Zymo Research (D7001, https://www.zymoresearch.com/collections/quick-dna-rna-kits/products/quick-dna-rna-miniprep). The key difference is that we find RNA binding buffer (R1013) is far more effective as a lysis buffer for our yeast samples than the buffer supplied with that kit. The protocol makes extensive use of Zymo Research columns and buffers, that can be purchased individually. Other manufacturers make similar columns and buffers or you can make your own, although we have not tested those (Yaffe et al. 2012; Jue et al., 2020).
References:
Jue, E., Witters, D. & Ismagilov, R.F. Two-phase wash to solve the ubiquitous contaminant-carryover problem in commercial nucleic-acid extraction kits. Sci Rep 10, 1940 (2020). https://doi.org/10.1038/s41598-020-58586-3
Yaffe, H., Buxdorf, K., Shapira, I. et al. LogSpin: a simple, economical and fast method for RNA isolation from infected or healthy plants and other eukaryotic tissues. BMC Res Notes 5, 45 (2012). https://doi.org/10.1186/1756-0500-5-45
Steps
Grow single colonies of yeast
Streak out yeast from glycerol stocks to single colonies on petri dishes poured with solid media that is appropriate for your experiment, and grow as desired. We usually grow for 2 days (48h 0m 0s) at 30°C. To minimize experimental variability, it is best to use fresh plates, i.e. that have never been stored in a fridge.
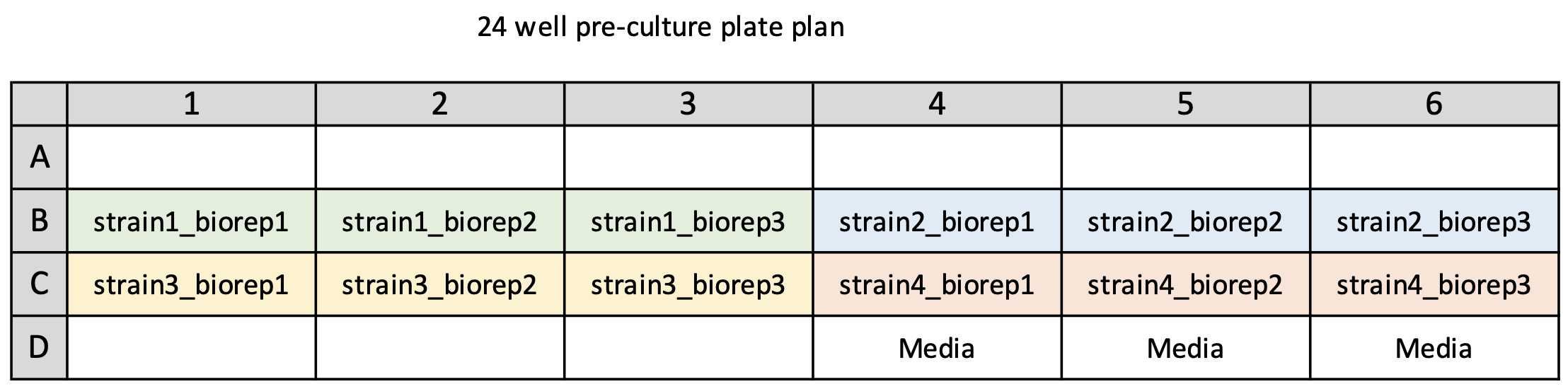
Plate plan
Plan your 24 well culture plate layout -
Subdivide strains into groups of 6 then fill out the strain names on the following pre-culture (24 well) plate plan to log the locations of each strain on the 24 well plate
Template 24 well plate plan - 24well_preculture_plateplan_template.xlsx
Example 24 well plate plan - 24well_preculture_plateplan_example1.xlsx
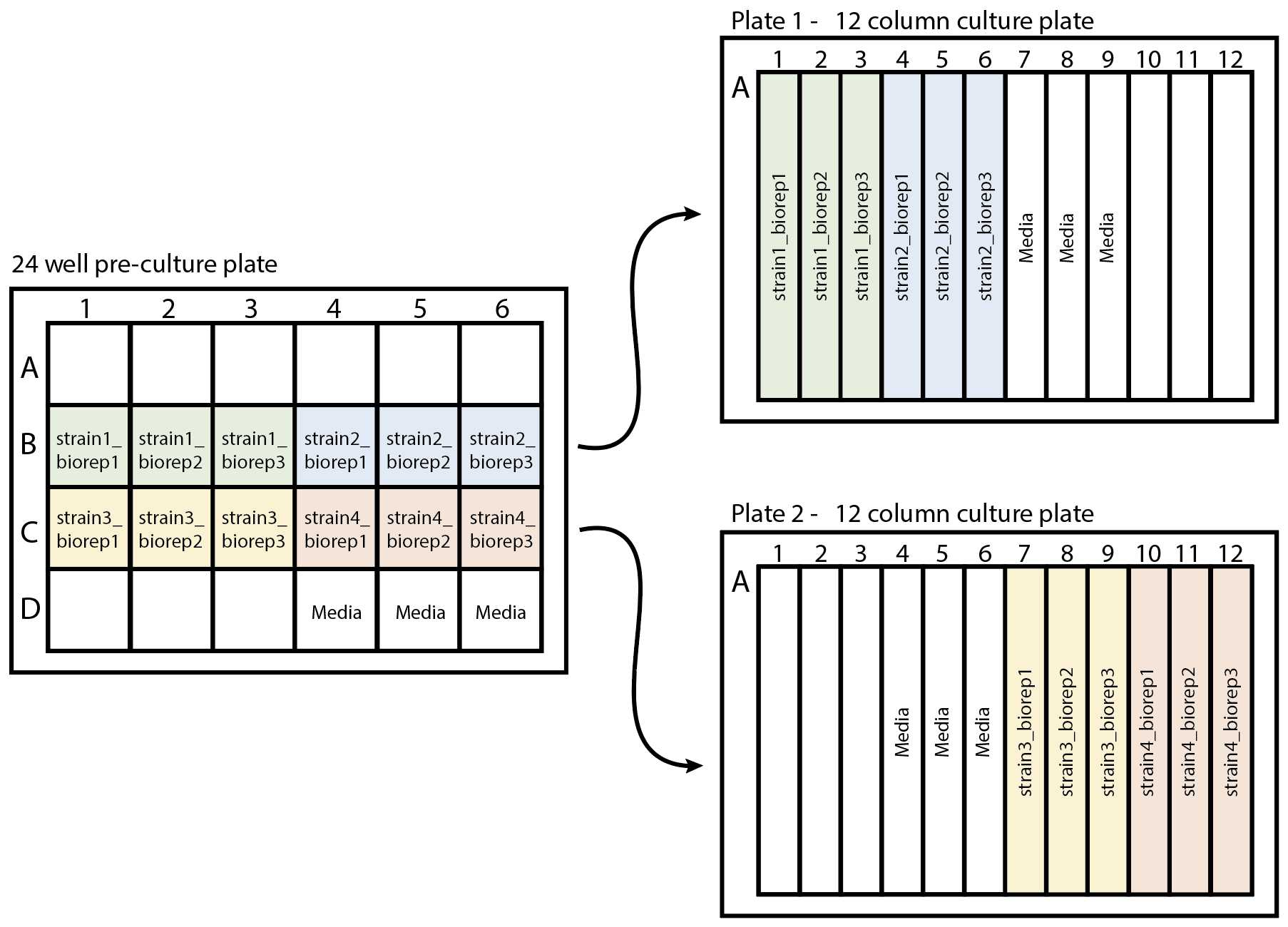
Plan your 12 column culture plate layout -
Subdivide the strains into groups of 12 then fill out the strain names on the following 12 well plate plan to log the locations of each strain on plate.
Template 12 column deep well plate plan - 12column_culture_plateplan_template.xlsx
Example 12 column deep well plate plan - 12column_culture_plateplan_example1.xlsx

Pre-culture growth
Take a sterile 24 (deep) well plate (Star lab cat no. E2824-0000) and using a pipette controller, transfer 1mL sterile media to each well of the 24 well plate according to the plate plan above (24 well plate plan in Step 2).
Using sterile P20 tips, inoculate the media in each well with a single colony of the appropriate strain according to the plate plan above (24 well plate plan in Step 2).
Seal the deep well plate with a gas permeable seal (Thermo cat #AB0718) and grow overnight in a shaking incubator set at 30°C and 200 rpm.
After 16-20 hours, for each strain transfer 100µL of the overnight culture to a cuvette then add 900µLof media and mix well (1:10 dilution of the overnight culture) and measure the OD600 using a spectrophotometer.
Calculate the amount of the overnight culture to add to 8 ml of sterile media to reach a starting OD600 of 0.2.
Culture growth from an OD600 of 0.2 to 0.5-0.7
Take sterile 12 column deep well plates (4titude cat #PCR0244) (based on the plate plans above, we will need 2 for this example). Using a pipette controller (Eppendorf cat #4430000018), dispense 7mL of media into each well of the 12 column deep well plate.
Transfer the calculated amount of the overnight culture into their respective wells on the 12 column deep well plate, based on the amount calculated in step 8.
Take 1mL of the diluted culture and measure their OD600 using a spectrophotometer (to check that their starting ODs are betwee 0.15 - 0.2).
Seal with a gas permeable seal and grow in a shaking incubator set at 30°C and 90 rpm to an OD600 of 0.5 - 0.7.
After ~3.5 - 4 hours, measure the OD of each culture by transferring 1mL of the culture using a spectrophotometer.
Harvesting and lysis of cells
Once the OD600 of the culture has reached between OD600 of 0.5-0.7, pellet the cells on the 12 column deep well plate by centrifugation at 3000x g.
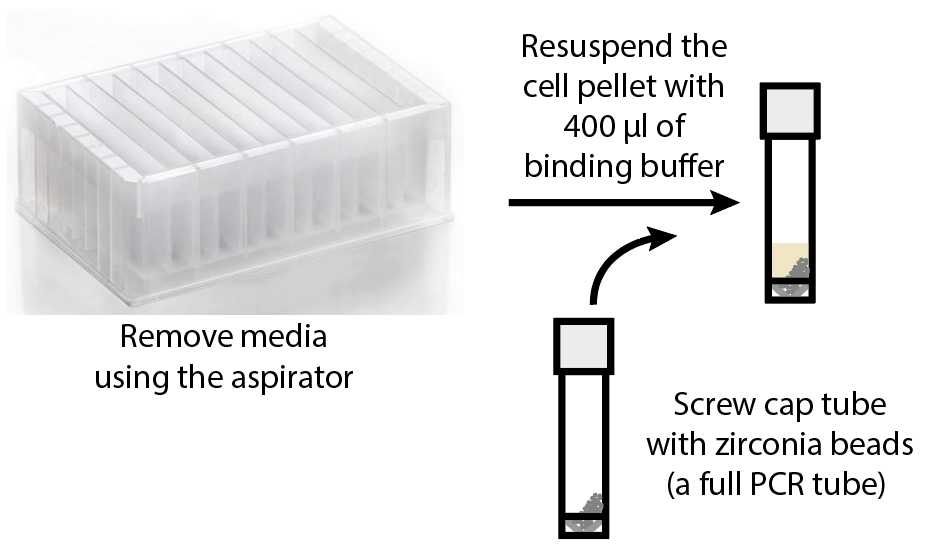
Using an aspirator, remove the media in each well. Avoid touching the pellet.
.")
Take a screw cap tube (Sarstedt cat #12705493) and use a PCR tube to measure zirconia beads (BioSpec #11079105z) (note - fill the entire PCR tube with beads) . Add the zirconia beads into the screw cap tubes. Label each tube.
Resuspend the pellet in the 12 column deep well plate with400µL of RNA binding buffer (Zymo cat no. R1013-2) and transfer them to the screw cap tubes containing the zirconia beads (keep on ice)
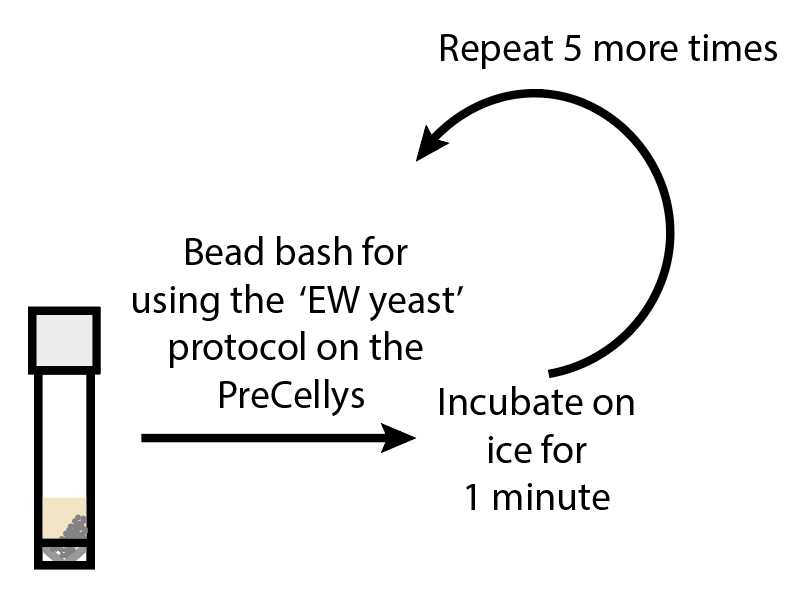
Transfer the tubes to the PreCellys (Bertin Instruments PreCellys Evolution) and run the EW Yeast proto c ol (this is a0h 0m 50s method). Chill tubes on ice for 0h 1m 0s . Repeat this step 5 more times (Total of 6 cycles)
The Precellys Evolution EWYeast protocol is set to 3 cycles shaking at a speed of 6000 RPM, each with a duration of 10s and with a pause between each cycle of 10s.
Transfer the tubes to a tabletop centrifuge and spin at 12000x g.
Transfer supernatant to a Zymo Spin IIICG column (Zymo cat no. C1006) on a collection tube (Label tubes). Spin the Zymo Spin IIICG containing the supernatant at 12000x g. (This step binds DNA to IIICG column).
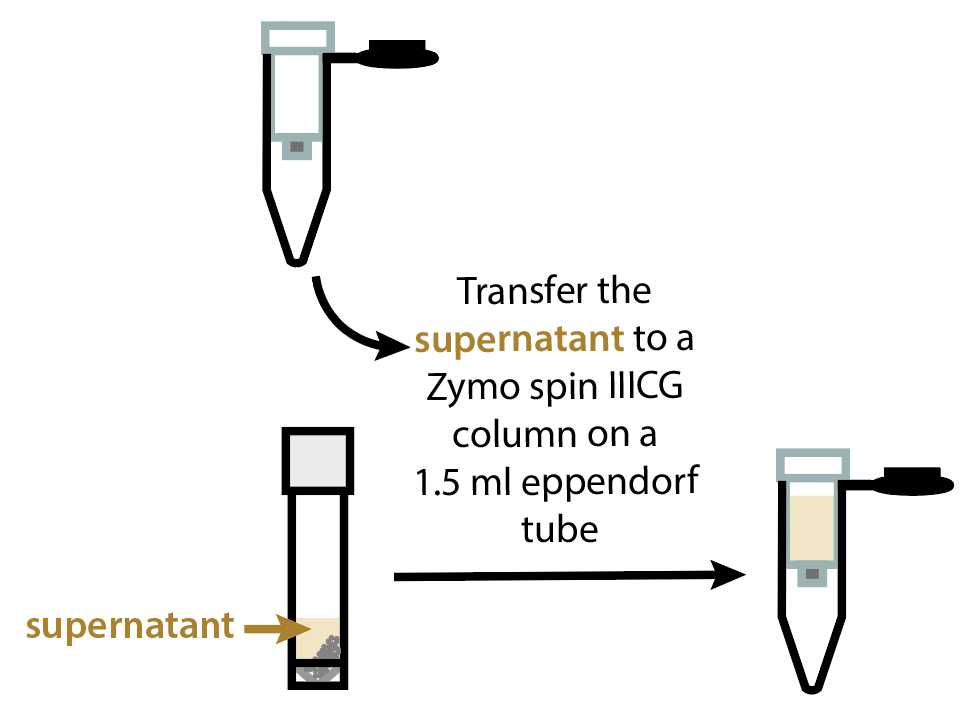
RNA extraction
Discard the Zymo IIICG and keep the flow through ! Add 400µL of 100% EtOH to the flow through on the collection tube and mix well by pipetting up and down at least 4x.
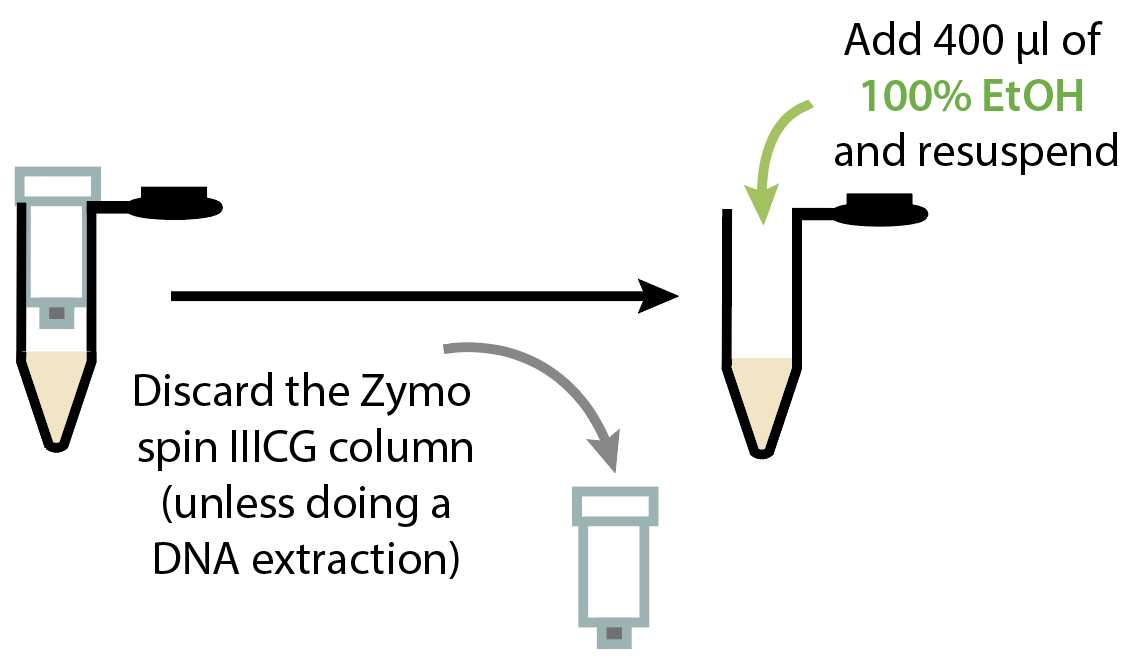
Transfer the flow-through and ethanol mixture to a Zymo Spin IIC column (Zymo cat no. C1011) on a collection tube and spin at 12000x g.
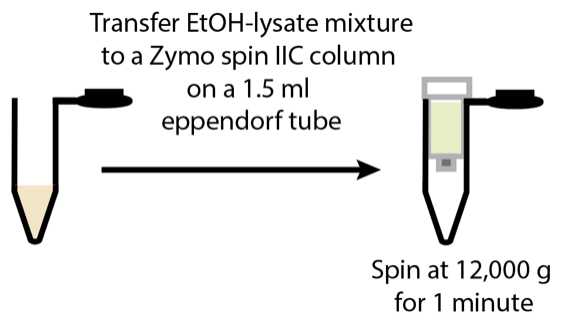
Discard the flow-through and keep the column. Add 400µL of DNA/RNA Prep Buffer (Zymo cat no. D7010-2) to the column. Spin at 12000x g.
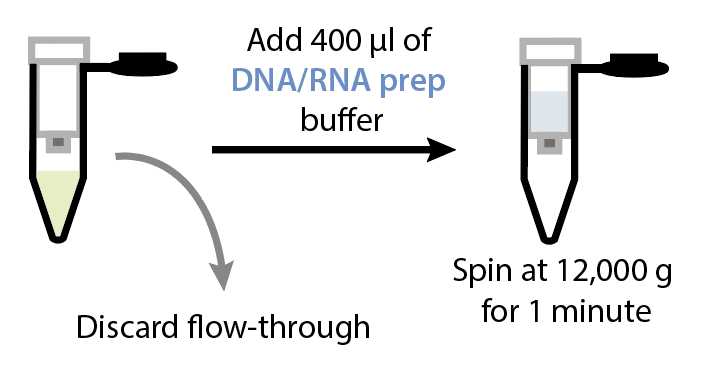
Discard the flow-through and add 600µL of DNA/RNA Wash Buffer (Zymo cat no. D7010-3) to the column. Spin at 12000x g.
Discard the flow-through and add 400µL of DNA/RNA Wash Buffer to the column. Spin at 12000x g.
Discard the flow-through spin again at 12000x g.
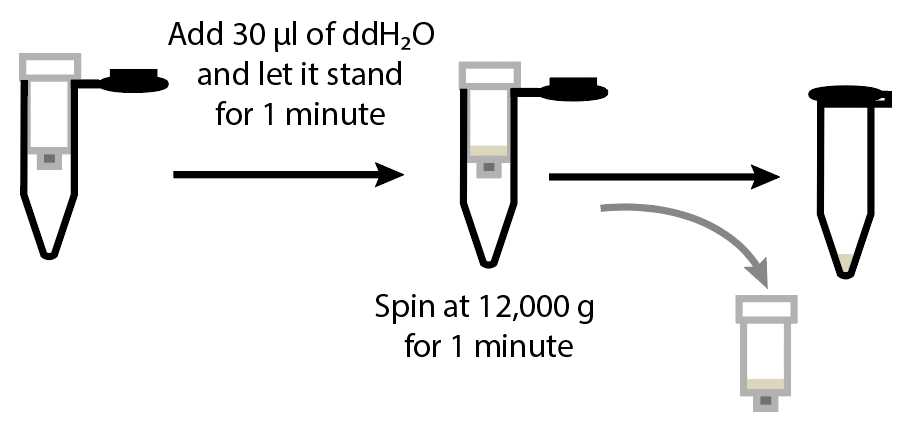
Transfer the Zymo IIC column to a clean 1.5 ml tube, labelled with your sample name as this will be used to store the RNA. Add 30µLof nuclease free water to the column (add this on the filter without touching the filter with your tip) and incubate at room temperature for 0h 1m 0s . Spin at 12000x g and discard the column. The RNA is now in the eluted flow-through.
Measure the concentration of the RNA using a microvolume spectrometer (e.g.nanodrop). Verify the quality of the RNA by running samples on the fragment analyzer.
