Preparation of viral sequencing library for Illumina using NEBNext ultra II
Kenichi Komabayashi
Disclaimer
DISCLAIMER – FOR INFORMATIONAL PURPOSES ONLY; USE AT YOUR OWN RISK
The protocol content here is for informational purposes only and does not constitute legal, medical, clinical, or safety advice, or otherwise; content added to protocols.io is not peer reviewed and may not have undergone a formal approval of any kind. Information presented in this protocol should not substitute for independent professional judgment, advice, diagnosis, or treatment. Any action you take or refrain from taking using or relying upon the information presented here is strictly at your own risk. You agree that neither the Company nor any of the authors, contributors, administrators, or anyone else associated with protocols.io, can be held responsible for your use of the information contained in or linked to this protocol or any of our Sites/Apps and Services.
Abstract
This method uses a metagenomic approach to analyze the genome sequence of RNA viruses. Nucleic acids outside the viral particles are reduced using nucleases and extracted to obtain template RNA. Templates are converted to double-stranded DNA, and library preparation is performed for analysis on Illumina sequencers.
Analysis data with reduced sequences of host and bacterial origin and abundant sequences of viral origin are obtained, allowing multiple samples to be analyzed even with the throughput of the iSeq100.
This protocol was folked from "Preparation of viral sequencing library for Illumina using WTA2 and QIAseq FX".
Steps
Reduction of nucleic acids derived from non-virus (pretreatment)
Collect 400µL virus culture medium in a 1.5 mL tube.
Centrifuge 0h 3m 0s at 17,000 x g and aspirate the supernatant with a 1 mL tuberculin syringe.
Equipment
| Value | Label |
|---|---|
| New Steradisc | NAME |
| 0.45μm filter 50pcs | TYPE |
| Kurabo | BRAND |
| S-1304 | SKU |
| https://www.kurabo.co.jp/english/ | LINK |
Filter the medium through a 0.45μm filter into a 1.5 mL tube.
Mix the following reagents in a new 1.5mL tube.
**Component Volume / sample**
Micrococcal nuclease `1µL`
Benzonase `2µL`
Homemade buffer* `7µL`
*see MATERIALS
Add 200µL of filtrate into the tube, then mix by pipetting.
Incubate at 37°C for 2h 0m 0s.
Extract RNA from total volume (210µL) and elute to 50µL.
Fragmentaion of RNA and priming
0.2 mL PCR tubes are used to incubate mixtures.
For steps 9 to 47, refer to section 2 'Protocol for use with NEBNext rRNA Depletion Kit v2 (Human/Mouse/Rat)' in a manual of NEBNext Ultra II RNA Library Prep kit for Illumina (kit E7770). In our protocol, section 2.5 and beyond is referred.
Half volume of the reagent listed in the manual is used.
Mix the following components, keep4On ice
**Component Volume / sample**
RNA `2.5µL`
First Strand Synthesis Buffer (kit E7770) `2.0µL`
Random Primers (kit E7770) `0.5µL`
Total so far: 5µL
Incubate in a thermal cycler set with the following program.
Keep the heat-lid at 105°C.
1.`94°C` for `0h 10m 0s`
2. Hold at `4°C`
Synthesis of 1st strand cDNA
Mix the following components, keepOn ice
Component Volume / sample
Product from step 10 `5.0µL`
First Strand Sysnthesis Enzyme (kit E7770) `1.0µL`
Nuclease Free Water `4.0µL`
Total so far: 10µL
Incubate in a thermal cycler set with the following program.
Keep the heat-lid at 80°C.
1. `25°C` for `0h 10m 0s`
2. `42°C` for `0h 50m 0s`
3. `70°C` for `0h 15m 0s`
4. Hold at `4°C`
Synthesis of 2nd strand cDNA
Mix the following components, keepOn ice
Component Volume / sample
Product from step 13 `10µL`
Second Strand Synthesis Buffer (kit E7770) `4.0µL`
Second Strand Synthesis Enzyme (kit E7770) `2.0µL`
Nuclease Free Water `24µL`
Total so far: 40µL
Incubate in a thermal cycler set with the following program.
Keep the heat-lid at 40°C.
1.`16°C` for `1h 0m 0s`
2. Hold at `4°C`
Clean-up using magnetic beads
Clean-up products using
Add 72µL (1.8x) of AMpure XP per sample.
Incubate atRoom temperature for 0h 5m 0s
Separate magnetic beads and remove supernatant.
To wash beads, add 200µL of 80% ethanol, incubate for 0h 0m 30s, and remove supernatant (1/2)
To wash beads, add 200µL of 80% ethanol, incubate for 0h 0m 30s, and remove supernatant (2/2)
Allow the beads to dry for 0h 2m 0s.
Elute purified product in 26µL of 0.1x TE (kit E7770).
Separate magnetic beads and transfer 25µL of supernatant to a new 0.2 mL tube.
End Prep of cDNA Library
Mix the following components, keepOn ice
**Component Volume / sample**
Product from step 13 `25µL`
End Prep Reaction Buffer (kit E7770) `3.5µL`
End Prep Reaction Enzyme (kit E7770) `1.5µL`
Total so far: 30µL
Incubate in a thermal cycler set with the following program.
Keep the heat-lid at 75°C.
1. `20°C` for `0h 30m 0s`
2. `65°C` for `0h 30m 0s`
3. Hold at `4°C`
Adaptor ligation
Mix the following components in a 1.5 mL low-binding tube, keep On ice
Component Volume
NEBNext Adaptor for Illumina (E7335 or E7500) `1.0µL`
Adaptor Dilution Buffer(kit E7770) `199µL`
Mix the following components as master mix in a 1.5 mL tube, keep On ice
Component Volume / sample
Ligation Enhancer (kit E7770) `0.5µL`
Ligation Master Mix (kit E7770) `15µL`
Mix the following components, in the order given, keep On ice
Component Volume / sample
Product from step 24 `30µL`
Diluted Adaptor (step 25) `1.25µL`
Master mix (step 26) `15.5µL`
Total so far: 46.75µL
Incubate in a thermal cycler set with the following program.
Keep the heat-lid at 45°C.
1. `20°C` for `0h 15m 0s`
2. `20°C` pose *
3. `37°C` for `0h 15m 0s`
4. Hold at `15°C`
- Add USER Enzyme (kit E7770)
1.5µL/ sample and mix
Total so far: 48.25µL
Clean-up products using
Add 43.5µL (0.9x) of AMpure XP per sample.
Incubate atRoom temperature for 0h 10m 0s
Separate magnetic beads and remove supernatant.
To wash beads, add 200µL of 80% ethanol, incubate for 0h 0m 30s, and remove supernatant (1/2)
To wash beads, add 200µL of 80% ethanol, incubate for 0h 0m 30s, and remove supernatant (2/2)
Allow the beads to dry for 0h 2m 0s.
Elute purified product in 8.0µL of 0.1x TE (kit E7770).
Separate magnetic beads and transfer 7.5µL of supernatant to a new 0.2 mL tube.
PCR Enrichment of Adaptor Ligated DNA
Mix the following components, keep On ice
Component Volume / sample
Adaptor Ligated DNA from step 36 `7.5µL`
Q5 Master Mix (kit E7770) `12.5µL`
Index (X) Primer (E7335 or E7500) `2.5µL`
Universal PCR Primer (E7335 or E7500) `2.5µL`
For multiplex analysis of specimens fewer than seven, use 'Index oligo selector' to verify that the index combination is acceptable.
Incubate in a thermal cycler set with the following program.
-
98°Cfor0h 0m 30s -
20 cycles x (
98°Cfor0h 0m 10s,65°Cfor0h 1m 15s) -
Hold at
4°C
Clean-up of PCR product using magnetic beads and quantification of DNA
Clean-up products using
Add 22.5µL (0.9x) of AMpure XP per sample.
Incubate atRoom temperature for 0h 5m 0s
Separate magnetic beads and remove supernatant.
To wash beads, add 200µL of 80% ethanol, incubate for 0h 0m 30s, and remove supernatant (1/2)
To wash beads, add 200µL of 80% ethanol, incubate for 0h 0m 30s, and remove supernatant (2/2)
Allow the beads to dry for 0h 2m 0s.
Elute purified product in 11.5µL of 0.1x TE (kit E7770).
Separate magnetic beads and transfer 11µL of supernatant to a new 0.2 mL tube.
Quantify the purified amplicon using fluorescent based method using
Concentrations in the range of 10-100 ng/µL of purified amplicon are sufficient for the next section.
Library pooling
Take the purified PCR product from each tube and pool them into the 1.5 mL low-binding tube.
Adjust the volume to be pooled to average the amount of DNA in each sample.
Briefly measure the volume of pooled mixture using pipette.
Add 0.1x TE (kit E7770) up to 50µLof total volume.
Purification of the library for size selection
Clean-up products using
Add 30µL (0.6x) of AMpure XP per sample.
Incubate atRoom temperature for 0h 5m 0s
Separate magnetic beads and remove supernatant.
To wash beads, add 200µL of 80% ethanol, incubate for 0h 0m 30s, and remove supernatant (1/2)
To wash beads, add 200µL of 80% ethanol, incubate for 0h 0m 30s, and remove supernatant (2/2)
Allow the beads to dry for 0h 2m 0s.
Elute purified product in 30µL of 0.1x TE (kit E7770).
Separate magnetic beads and transfer supernatant to a new 0.2 mL tube.
Quantify the purified amplicon using fluorescent based method using
Concentrations 1.5 ng/µL or more of purified library is sufficient for the next section.
Estimation of library size
Quantify the purified library using
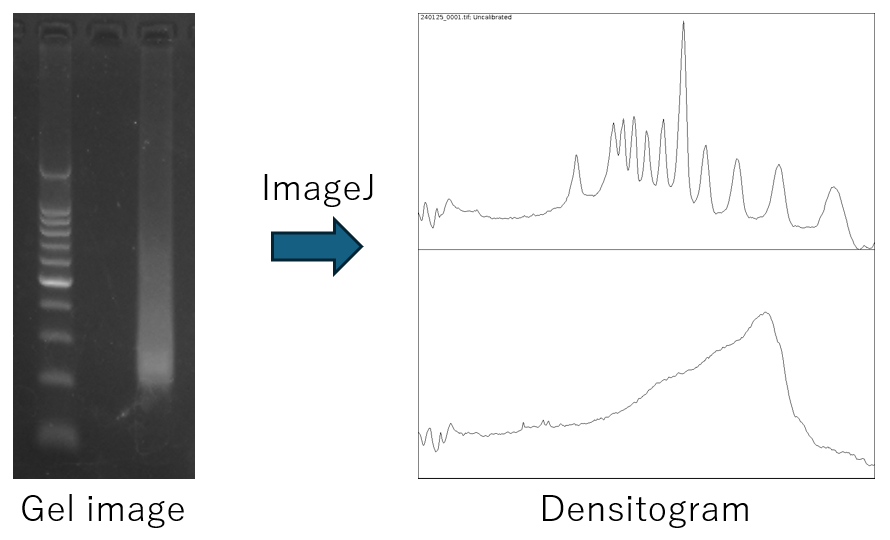
Mix 5µL of the library with loading dye and electrophoresis on a 2% agarose gel alongside molecular markers.
Obtain a smear image of the library.
Estimate approximate average library size (base pairs) on the smear image.
The size of the most concentrated region can be read and used as an estimation.
Preparation of 50pM library for Illumina iSeq100
Calculate molar concentration of the library using the formula below.
Y (nM) = X (ng/µL) ÷Z (base pairs) ÷ 660 (g/mol) ×106
Y: molar concentration of the library
X: mass concentration of the library
Z: average library size
Setting up the local run manager in iSeq100
To analyze libraries using NEBNext® multiplex oligos for Illumina, you need to load the index and other information into a local run manager.
Refer to the manuals on Illumina site
How to use a custom library prep kit in Local Run Manager v2
How to use custom library prep and index kits with Local Run Manager v3 and v4
Obtain a .tsv file for configuration from the NEBNext® multiplex oligo page on NEB.
Change DefaultReadLength1, 2 in the .tsv file from 251 to 151.
Start the Local Run Manager on iseq100 and open "Tools" on the dashboard.
In the drop-down menu, select "Index & Library Prep Kits", "Index Kit", and "Add Index Kit".
Select and load the modified .tsv file.
