High Molecular Weight Total DNA Extraction from plant tissues for Long Read Sequencing
Subash Rai
High Molecular Weight DNA
Plant HMW DNA extraction
Neptunia HMW DNA
long read sequencing
HMW total DNA
Abstract
This protocol was developed as a research within GIH collaborative projects for a sample where a species of Neptunia leaves sample was problematic in our previously developed protocol. At step 21 of previous protocol, the solution forms either a brownish mark on the top layer of the solution or the whole solution depending on the starting sample amount which resulted brown CTAB-DNA complex. In this protocol, the problematic steps for a particular sample is improved to extract high quality High Molecular Weight (HMW) DNA >60kb. The DNA quality was assessed in Qubit, NanoDrop, TapeStation, and Oxford Nanopore Technologies. Using a LSK109 ligation chemistry and R9.4 flow cell, a total yield of 24gb with N50 29kb was generated in MinION sequencing platform.
Before start
Prepare the following buffers and solutions before starting the experiment:
Lysis Buffer
| A | B |
|---|---|
| Tris-HCl | 100 mM |
| EDTA | 20 mM |
| CTAB (w/v) | 4% |
| NaCl | 1.4 M |
| PVP 360k (w/v) | 1% |
| β-mercaptoethanol (add just before use)] | 2% |
Combine the reagents given in the table below.
| A | B |
|---|---|
| 1M Tris-HCl (pH = 8) | 5 ml |
| 0.5M EDTA (pH = 8) | 2 ml |
| CTAB powder | 2 g |
| PVP | 0.5 g |
| NaCl | 4 g |
Adjust the final volume to 50mL with Nuclease free water/lab grade water. Store at -80Room temperature for up to 3-4months.
High-salt TE buffer
| A | B |
|---|---|
| EDTA | 2 mM |
| Tris-HCl | 10 mM |
| NaCl | 1 M |
Combine the reagents given in the table below
| A | B |
|---|---|
| NaCl | 581 mg |
| 0.5M EDTA (pH=8) | 40 μl |
| 1M Tris-HCl (pH=8) | 100 μl |
Complete to 10mL with Distilled water Ultra-Pure. Autoclave it for long-term (1 year) storage.
Binding buffer (20% PEG8000 and 3M NaCl):
Add 2g PEG 8000 and 1.75g NaCl in 10mL nuclease free water and mix well until it turns as a clear solution and store at cold room or 4-7°C.
Beads solution :
| A | B |
|---|---|
| Dynabeads™ M-270 Carboxylic Acid | 4% |
| PEG8000 | 18% |
| NaCl | 1 M |
| Tris-HCl pH-8 | 10 mM |
| EDTA pH-8 | 1 mM |
- First prepare the required volume of the solution except Dynabeads.
- Keep the Dynabeads at RT for at least
0h 15m 0s. Mix well by vortexing, then take 4% of the beads solution (v/v) immediately. - Wash the beads with nuclease free water 3 times. Resuspend the beads pellet completely while washing.
- Add the beads solution and store the beads solution at
4°C. - Keep the beads solution at
4Room temperaturefor at least0h 15m 0sand mix well before using it.
Attachments
Steps
Tissues preparation and lysis
Take 10mL lysis buffer and warm it at 60°C for 15-20 min.
Take ~1L of liquid nitrogen (LN2) in Dewar Flask that requires for chilling mortar and pestle and grinding the tissues.
Take dry ice in an esky/insulated container for later steps.
Grind 500mg to 1000mg healthy young fresh/snap frozen/frozen tissues in mortar and pestle chilled with LN2 to fine powder.
Keep a 15 ml falcon tube on the dry ice for 0h 5m 0s then swirl the ground powder with LN2 and pour directly into the falcon tube while keeping the falcon tube on the dry ice.
Keep the lid half-opened and let LN2 to evaporate.
Take out the tube and add 10mL prewarmed lysis buffer (at 60°C) with freshly added 200µL β- mercaptoethanol.
Mix well by inverting the tubes (~100 times) until the solution become more homogenous. In some sample, solution may not be homogenous but form whiteish clumps (it is normal) and incubate at 60°C in thermomixer at 300rpm,0h 0m 0s for 0h 30m 0s.
Add 200µL Proteinase K (stock conc=20mg/mL) after 15 min of incubation.
Mix well by inverting the tube (15-20 times) and continue the incubation.
Spin the solution at 3000x g.
Take an equal volume of the supernatant in two fresh 15 ml falcon tubes using P1000 wide bore pipette tips.
Extraction of raw HMW DNA
Add an equal volume of Chloroform:Isoamyl alcohol (24:1) into the solution.
Mix the solution by inverting the tube until a milky colour appears (~100 times) and centrifuge at 3000x g.
Transfer the aqueous phase to a new 15ml falcon tube without disturbing interface layer.
Add an equal volume of Chloroform:Isoamyl alcohol (24:1) into the solution.
Mix the solution by inverting the tubes ~100 times and centrifuge at 3000x g.
Transfer 1 ml aqueous phase to 2 ml LoBind tube without disturbing the interface layer (much thinner than the first extraction). It requires multiple 2 ml LoBind tube.
Add half volume of Ammonium acetate (7.5 M). Mix well by inverting the tubes and incubate for 0h 10m 0s at 60Room temperature
Centrifuge at 13000x g .
Transfer the supernatant in a fresh 2ml LoBind tube and add equal volume of Isopropanol (>98%), mix well, and incubate for 0h 10m 0s at Room temperature .
Resuspend the pellet with 1mL 70% ethanol ( freshly prepared ) using a wide bore P1000 pipette tip and transfer all into a 2ml LoBind DNA tube.
Rinse the tube with additional 1mL 70% ethanol to collect remaining CTAB-DNA complex. Perform the same for another 15 ml falcon tube.
Incubate 2 ml tubes for 0h 5m 0s at 60Room temperature in a HulaMixer at 9rpm,0h 0m 0s.
Spin the tube at 13000x g and discard the supernatant.
Repeat washing steps & once with 2mL 70% ethanol.
Keep the tubes under fume hood for 0h 5m 0s to remove any traces of ethanol.
Resuspend the DNA pellet in 200µL of prewarmed (60°C) High-salt TE buffer .
Add 4µL RNaseA and incubate at 37°C for 15-20 min.
Beads Purification of HMW DNA
Add 100µL ammonium acetate (7.5Molarity (M)) mix well and incubate it for 0h 10m 0s at 37Room temperature. Shake it once every 5 min.
Spin the tube at 13000x g and transfer the supernatant using wide-bore pipette tips into a fresh tube.
Add equal volume of binding buffer and mix well by inverting the tube.
Add 150µL beads solution (8-9 million beads) and incubate it for 0h 30m 0s at 37Room temperature in a HulaMixture.
Place the tube in magnetic rack for 2-3 min and remove the supernatant and wash the beads with 500µL freshly prepared 70% ethanol (clumping of beads may appear but try to dislodge by inverting the tube several times).
Wash the pellet once again with 500µL freshly prepared 70% ethanol.
Take out the tubes from magnetic rack and add 500µL freshly prepared 70% ethanol.
Dislodge the beads by inverting the tubes.
Place the tubes back to the magnetic rack for 2 min and remove the supernatant.
Add 75µL prewarmed (at 50°C) 10millimolar (mM) Tris-HCl (elution buffer) 8 and incubate at 37Room temperature for 0h 15m 0s.
Place the tube back in the magnetic rack and leave it for 0h 5m 0s.
Remove the supernatant in the fresh 1.5ml LoBind DNA tube.
Assess DNA quality in NanoDrop, Qubit, and TapeStation/PFGE
Worked Results
General Quality check:
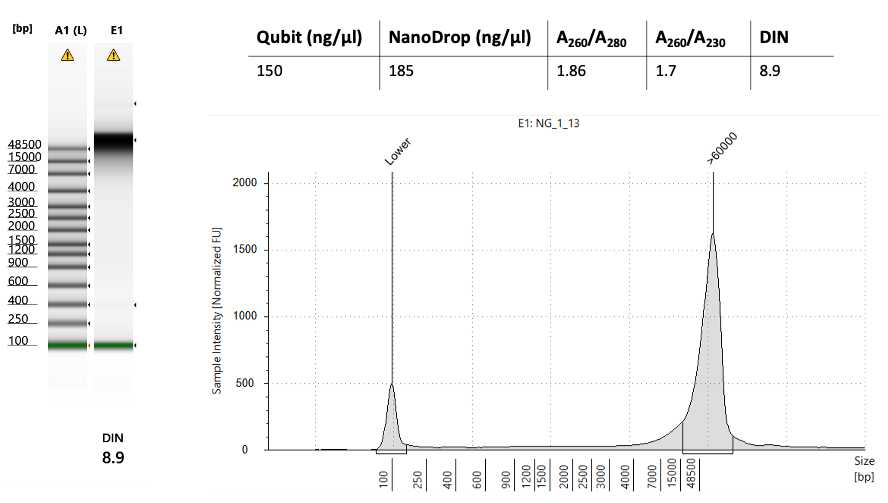
QC check in Oxford Nanopore Sequencing:
 in MinION Nanopore sequencing. DNA was extracted from frozen leaves sample. The size selection was performed with standard SRE kit (circulomics) prior to the library preparation using ONT LSK109 kit.")