FIND-seq protocol v1.0
Iain C. Clark, Michael A. Wheeler, Hong-Gyun Lee, Liliana M. Sanmarco, Zhaorong Li, Shravan Thaploo, Carolina M. Polonio, Seung Won Shin, Giulia Scalisi, Joseph M. Rone, Federico Giovannoni, Stephanie E. J. Zandee, Alexandre Prat, Daniel C. Douek, Eli A. Boritz, Francisco J. Quintana, Adam R. Abate
Single cell sequencing
RNA sequencing
droplet microfluidics
droplet cytometry
nucleic acid cytometry
Disclaimer
DISCLAIMER – FOR INFORMATIONAL PURPOSES ONLY; USE AT YOUR OWN RISK
The protocol content here is for informational purposes only and does not constitute legal, medical, clinical, or safety advice, or otherwise; content added to protocols.io is not peer reviewed and may not have undergone a formal approval of any kind. Information presented in this protocol should not substitute for independent professional judgment, advice, diagnosis, or treatment. Any action you take or refrain from taking using or relying upon the information presented here is strictly at your own risk. You agree that neither the Company nor any of the authors, contributors, administrators, or anyone else associated with protocols.io, can be held responsible for your use of the information contained in or linked to this protocol or any of our Sites/Apps and Services.
Abstract
This protocol is a detailed description of FIND-seq, a single cell method for sorting cells based on RNA or DNA biomarkers. The protocol contains step-by-step instructions, key checkpoints, and troubleshooting guidelines.
Steps
REAGENT SETUP
20% (vol/vol) 1H-1H-2H-2H-Perfluoro-1-Octanol solution (Use this reagent immediately after preparation.)
| A | B |
|---|---|
| Reagent | Vol. |
| PFO | 10 mL |
| HFE-7500 | 40 mL |
| Total | 50 mL |
0.1% SPAN-80 in Hexane (Use this reagent immediately after preparation.)
| A | B | C | D |
|---|---|---|---|
| Reagent | Reagent conc. | Final conc. | Vol. |
| Span-80 (in hexane) | 20% (w/v) | 0.1 % (w/v) | 0.25 mL |
| n-hexane | - | - | 50 mL |
| Total | 50.25 mL |
Cell Resuspension Buffer ( Solution is stable for several days at 4°C.)
| A | B | C | D |
|---|---|---|---|
| Reagent | Reagent conc. | Final conc. | Vol. |
| HBSS buffer | 1X | - | 7.2 mL |
| Pluronic F-68 | 10% (v/v) | 1% (v/v) | 1 mL |
| Opti-prep | - | 18% (v/v) | 1.8 mL |
| Total | 10 mL |
Lysis Buffer ( This solution is stable for several months at -20°C and should be stored in small aliquots. When needed for experiment, add Proteinase K (2 µg/µL) to an aliquot and use immediately after preparation.)
| A | B | C | D |
|---|---|---|---|
| Reagent | Reagent conc. | Final conc. | Vol. |
| Tris-HCl pH 7.5 | 1000 mM | 20 mM | 0.2 mL |
| LiCl | 8000 mM | 1000 mM | 1.25 mL |
| LiDS | 10 % (v/v) | 1 % (v/v) | 1 mL |
| EDTA | 500 mM | 10 mM | 0.2 mL |
| DTT | 1000 mM | 10 mM | 0.1 mL |
| Proteinase K | 20 µg/µL | 2 µg/µL | 1 mL |
| Nuclease free water | - | - | 6.25 mL |
| Total | 10 mL |
Wash 1 Buffer (This solution is stable for several months at 4°C.)
| A | B | C | D |
|---|---|---|---|
| Reagent | Reagent conc. | Final conc. | Vol. |
| Tris-HCl pH 7.5 | 1000 mM | 20 mM | 10 mL |
| LiCl | 8000 mM | 500 mM | 31.25 mL |
| LiDS | 10% (v/v) | 0.1% (v/v) | 5 mL |
| EDTA | 500 mM | 0.1 mM | 0.1 mL |
| Nuclease free water | - | - | 453.65 mL |
| Total | 500 mL |
Wash 2 Buffer (This solution is stable for several months at 4°C.)
| A | B | C | D |
|---|---|---|---|
| Reagent | Reagent conc. | Final conc. | Vol. |
| Tris-HCl pH 7.5 | 1000 mM | 20 mM | 10 mL |
| NaCl | 5000 mM | 500 mM | 50 mL |
| Nuclease free water | - | - | 440 mL |
| Total | 500 mL |
5X RT Buffer (This solution is stable for several months at 4°C.)
| A | B | C | D |
|---|---|---|---|
| Reagent | Reagent conc. | Final conc. | Vol. |
| Tris-HCl pH 8.3 | 1000 mM | 250 mM | 50 mL |
| KCl | 1000 mM | 375 mM | 75 mL |
| MgCl2 | 1000 mM | 15 mM | 3 mL |
| DTT | 1000 mM | 50 mM | 10 mL |
| Nuclease free water | - | - | 62 mL |
| Total | 200 mL |
Tween Wash Buffer (This solution is stable for several months at room temperature.)
| A | B | C |
|---|---|---|
| Reagent | Final conc. | Vol. |
| Tween-20 | 0.1% (v/v) | 0.5 mL |
| Nuclease free water | - | 500 mL |
| Total | 500.5 mL |
Conjugation Buffer (Use this reagent immediately after preparation.)
| A | B | C | D |
|---|---|---|---|
| Reagent | Reagent conc. | Final conc. | Vol. |
| Tris-HCl pH 8.3 | 1000 mM | 375 mM | 7.5 mL |
| Nuclease free water | - | - | 12.5 mL |
10% (wt/vol) APS (Use this reagent immediately after preparation.)
| A | B | C |
|---|---|---|
| Reagent | Final conc. | Amount |
| APS | 10% (w/v) | 0.1 g |
| Conjugation buffer | - | upto 1 mL |
| Total | 1 mL |
10% (vol/vol) TEMED ( Use this reagent immediately after preparation.)
| A | B | C |
|---|---|---|
| Reagent | Final conc. | Vol. |
| TEMED | 10% (v/v) | 100 µL |
| Conjugation buffer | - | 900 µL |
| Total | 1000 µL |
( Caution: TEMED is toxic. Addition of TEMED should be done under a chemical fume hood. Wear appropriate protective clothing and equipment when handling it. )
AGAROSE CONJUGATION
Prepare reagents for agarose conjugation. Resuspend Acrydite-T5-Smart-dT Primer in Conjugation Buffer to a concentration of 1000micromolar (µM) (1millimolar (mM) ). Resuspend SFR Allyl Agarose in Conjugation Buffer in a 15 mL falcon to a final concentration of 0.5Mass / % volume. Prepare 10Mass / % volume APS and 10% (v/v) TEMED solutions.
Heat SFR Agarose suspended in buffer to 95°C for 2h 0m 0s, or until completely molten. Vortex over time to ensure homogenization. While vortexing, it is good to flip the tube and vortex on head/cap as well. This ensures agarose near the top of the falcon does not cool and harden.
Once homogenized, cool agarose to 45°C. Temporarily placing heat block in ice is used to accomplish this faster.
Place agarose under vacuum for 0h 30m 0s. Ensure the agarose does not boil. If boiling is seen, agarose is not cool enough.
Remove vacuum. Add reagents in the following order:
a. Add resuspended primer so final concentration of primer is 50micromolar (µM).
b. Add 10Mass / % volume APS solution so the final concentration is 0.1Mass / % volume. Vortex tube thoroughly.
c. Add 10% (v/v) TEMED solution so the final concentration is 0.1% (v/v). Volume of APS and TEMED added should be the same. Vortex tube thoroughly.
Place tube back under vacuum for 4h 0m 0s. PAUSE POINT
Remove tube from vacuum. Add the same volume of APS and TEMED as added in step 7. Final concentrations should now be 0.2Mass / % volume APS and 0.2% (v/v) TEMED. Vortex agarose and place under vacuum overnight at 45°C. PAUSE POINT
Remove agarose from vacuum. Heat to 95°C and vortex until completely molten and homogenized. Try to break polymer strands floating around by heating and vortexing as much as you can. Some strands may remain. While vortexing, it is good to flip the tube and vortex on the head/cap as well. This ensures agarose near the top of the tube does not cool and harden.
While molten, pour agarose in a 10 mL syringe with a 0.45 µm syringe filter attached. Filter agarose into another 15 mL falcon tube to remove unwanted polymer strands formed during the reaction.
Add ultra-low gelling temperature agarose to reach a final concentration of 2Mass / % volume agarose (SFR and Ultra-low gelling together).
After addition, heat tube to 95°C and vortex to ensure homogenization, flipping tube on head occasionally.
Once completely molten and homogenized, briefly centrifuge agarose to get all the agarose at the bottom of tube. Cool agarose to 4°C by placing in ice bucket for at least 1h 0m 0s. Allow agarose to harden.
Using a hypodermic needle, carefully pierce the bottom of the falcon tube. This should dislodge the hardened agarose from the bottom of the tube. Wash by transferring hard agarose gel to a 500 mL bottle of nuclease free distilled water and allow to sit overnight. PAUSE POINT
Repeat wash by re-transferring after the next night. Transfer agarose from wash bottle to a clean, dry 15 mL falcon tube. Ensure no water is transferred.
Agarose Normalization
Quantify the oligonucleotide concentration
Melt agarose at 95°C. Vortex and homogenize. Once agarose is molten, cool down to 70°C or until viscosity is amenable to pipetting. Use ice to cool agarose tube down.
Dilute agarose in nuclease free water (1/80 dilution). Vortex to ensure agarose added is dissolved and evenly distribute. Triplicates are ready for measurement.
In triplicate, measure conjugated oligo-dT via the QuBit ssDNA Assay kit.
Calculate the average µM concentration of conjugate oligonucleotide. Use the reported molecular weight on the IDT datasheet for the oligo-dT primer. Remember to consider the 80-fold dilution.
- e.g. If I get 5.52 ng/µL Qubit reading (1/80 dilution) for a 30 base ssDNA primer:
- Molecular concentration (µM) = Qubit reading (ng/µL) * Dilution factor (80) * 1000 / Molecular weight of total primer (g/mol)
- Molecular concentration (µM) = 5.52 ng/ul * 80 * 1000 / 9378.2 g/mol = 39 µM
Normalize the agarose concentration
Normalize the conjugated oligonucleotide concentration by addition of 2Mass / % volume Ultra-low melt Agarose. Prepare 2Mass / % volume Ultra-low melt Agarose by adding 0.08g of agarose to 4mL of distilled nuclease free water. Vortex and heat to 95°C. Keep vortexing until completely dissolved.
Dilute conjugated agarose in 2Mass / % volume Ultra-low melt Agarose to reach a final concentration of 8micromolar (µM) conjugated oligonucleotide.
Measure final conjugated oligonucleotide concentration with QuBit ssDNA kit using the same method as above. The final concentration should be 8micromolar (µM).
DAY 1: CELL ENCAPSULATION AND LYSIS
Heat 3mL of conjugated agarose-Oligo dT to 95°C for at least 1h 0m 0s. Vortex repeatedly to ensure homogenization and no solid clumps. CRITICAL STEP : Agarose must remain molten while running the device. Allowing agarose to cool down will clog the tubing/microfluidic channels.
Prepare cells for encapsulation. It is recommended to start with at least 25-30 million cells.
Preparing cells from cell lines in culture
Wash cells 1x in HBSS 400x g. Resuspend in 15mL HBSS for each wash. Filter through a 70-micron strainer.
Preparing PMBCs
a. Place 25mL of RPMI with 10% volume FBS in 37°C for 1h 0m 0s.
b. Remove PBMC vial from freezer on dry ice. ( CAUTION: Wear protective PPE, including a face shield, while defrosting PMBC vials. )
c. Submerge in 37°C water bath until only a small amount of ice is visible
d. Pipette cells into media that was pre-warmed in 37°C
e. Centrifuge cells for 300x g.
Count cells using Trypan blue stain. For cell lines, you can use an automated cell counter (Countess II Automated Cell counter). For PBMCs, we recommend a manual hemocytometer.
Based on the cell concentration, spin cells and resuspend in Cell Resuspension Buffer to 6.11x106 cells/mL. We recommend resuspending in smaller volume of Opti-Prep than necessary to obtain a higher concentration, and then diluting down to the required concentration.
*Troubleshooting
Setup the drop making station
Keep the agarose molten and place the bubble triggered co-flow microfluidic device on the stage. Connect the syringe tubing to the microfluidic device.
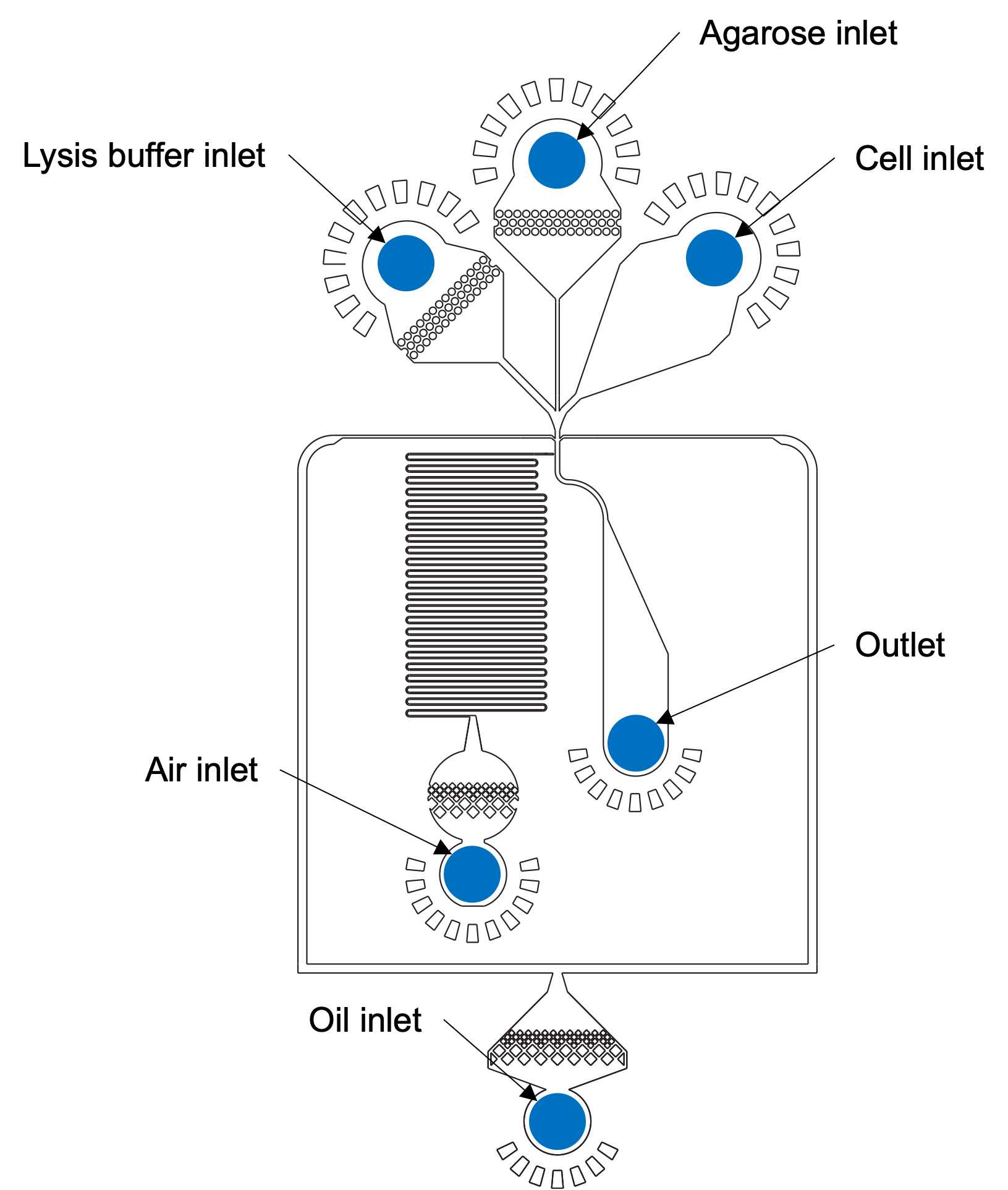
Encapsulate cells in molten agarose using the bubble triggered co-flow microfluidic device at the following flow rates.
| A | B | C |
|---|---|---|
| Syringe Size | Reagent | Final Flow Rate |
| 10 mL | Droplet Generation Oil for Probes (BioRad). | 5000 µL/hr |
| 3 mL | Cells filtered and resuspended in Cell Resuspension Buffer | 600 µL/hr |
| 3 mL | Lysis Buffer with Proteinase K | 600 µL/hr |
| 3 mL | Oligo-dT conjugated Agarose | 1200 µL/hr |
| - | Pressured air | 20 psi |
Collect drops in 15 mL tubes in the heat block at 55°C.
Incubate for 2h 0m 0s at 55°C. Cool on ice or at 4°C for at least 1h 0m 0s or overnight. PAUSE POINT
DAY 2: BREAKING AND REVERSE TRANSCRIPTION
Remove oil and wash with hexane ( Caution: Hexane and PFO are highly flammable substances. )
Hardened agarose is henceforth referred to as beads. Remove oil at bottom of tube. Discard oil in appropriate waste bottle.
Add 2x-5x volume 20% volume HFE/PFO solution to drop emulsion. Mix slowly by inverting tube by hand to adequately break emulsion.
Centrifuge at 2000x g. Oil will be at the bottom. Remove oil. Discard oil in waste bottle.
Add >25mL of Hexane/SPAN-80 solution to the beads. Shake tubes gently until beads are not clumpy. *Troubleshooting
Centrifuge at 2000x g. Remove hexane with a pipette and discard in a hexane waste container. ( CAUTION: Hexane damages plastics and should not be aspirated. Hexane must be disposed of in a glass waste bottle. )
CRITICAL STEP: You must proceed through the reverse transcription reaction. Beads must be kept on ice at 4°C throughout washes. Warming the beads will allow mRNA to dissociate from oligo-dT resulting in loss of mRNA and single cell resolution.
Water Washes
Add up to 50mL of 4°C Wash 1 Buffer. Resuspend by rotating by hand, flicking the bottom of the tube if necessary. Mix thoroughly and allow to sit on ice for 0h 5m 0s.
CRITICAL STEP : Residual hexane will be on top after first wash. Be careful to aspirate it and not let it mix in with the rest of the solution.
Centrifuge for 4500x g. Aspirate buffer, not beads. The largest source of bead loss is getting too close to the water-agarose interface.
Add up to 50mL Wash 2 Buffer. Resuspend by rotating by hand, flicking the bottom of the tube if necessary. Mix thoroughly and allow to sit on ice for 0h 5m 0s.
Pre-weigh a new 50 ml tube. Filter beads through 100 micron strainer into pre-weighted tube. *Troubleshooting
CRITICAL STEP : Pre-weighing the tube now is necessary for correctly setting up the reverse transcriptase reaction later in the protocol. Pre-weighed tube is used to calculate weight, and subsequently volume, of agarose beads left after washes.
Centrifuge for 4500x g. Aspirate buffer, not beads.
Add up to 50mL Wash 2 Buffer. Resuspend by rotating by hand, flicking the bottom of the tube if necessary. Mix thoroughly and allow to sit on ice for 0h 5m 0s.
Centrifuge for 4500x g. Aspirate buffer, not beads
Add up to 50mL 5X RT Buffer. Resuspend by rotating by hand, flicking the bottom of the tube if necessary. Mix thoroughly and allow to sit on ice for 0h 5m 0s.
Repeat wash (steps 36 to 38) for a total of 2 RT Buffer washes.
Reweigh tube after final aspiration to obtain weight of agarose beads.
Reverse Transcription Reaction
Beads should be in 5X RT Buffer on ice. Adding enzyme last, prepare reverse transcription reagents in a separate 50 mL falcon tube on ice as follows. Based on volume of washed beads, prepare reverse transcription mix.
| A | B | C | D |
|---|---|---|---|
| Reagent | Reagent conc. | Final conc. | Vol. |
| dNTP | 25 mM | 1 mM | 1.2 mL |
| TSO | 100 µM | 2 µM | 0.6 mL |
| MgCl2 | 1000 mM | 6 mM | 0.18 mL |
| Betaine | 5 M | 1 M | 6 mL |
| PEG-8000 | 30% (w/v) | 7.5% (w/v) | 7.5 mL |
| Maxima H minus Reverse transcriptase | 200 U/µL | 2 U/µL | 0.3 mL |
| NxGen Rnase inhibitor | 20 U/µL | 0.5 U/µL | 0.75 mL |
| Nuclease free water | - | - | 13.47 mL |
| Total | 30 mL |
Mix RT reaction using rotator for 0h 30m 0s at room temperature and then 1h 30m 0s at 42°C on a rotator.
Take out of incubator, add 200µL EDTA per 10mL of reaction. Cool on ice for 0h 10m 0s. PAUSE POINT
Alternatively, you can immediately begin bead washes with Tween Wash Buffer (next step).
Wash beads 5x in Tween Wash Buffer. On the last wash, filter with 100 micron strainer into a pre-weighed 50 mL Falcon tube.
After the final wash with Tween Wash Buffer, weigh to determine the final mass of beads.
Spin down beads for bead counting and take a 50µL aliquot. PAUSE POINT
Bead counting
Add 50µL of beads to 150µL of distilled nuclease free water to create a diluted bead stock.
In a separate tube, mix a small aliquot of beads from the diluted bead stock with SYBR green (10x final concentration of dye). Let sit for 0h 30m 0sin the dark.
Using hemocytometer, image beads fluorescence microscope. Take pictures of bead size, bead counts, and pictures at 20x magnification of bead lysis success.

Quantify the number of beads/µL and genomes/µL. The ratio of genomes/bead should be ~1/10.
Whole Transcriptome Amplification
Set up a 25µL PCR reaction as follows, thermocycling using three different cycle numbers (14, 16, 18). PAUSE POINT
| A | B | C | D |
|---|---|---|---|
| Reagent | Reagent conc. | Final conc. | Vol. |
| Kapa HiFi Master Mix | 2 X | 1 X | 12.5 µL |
| SMART PCR Primer | 10 µM | 0.4 µM | 1 µL |
| Beads | 30 genomes/µL | X µL | |
| Nuclease free water | - | - | 11.5 -X µL |
| Total | 25 µL |
| A | B | C | D | E |
|---|---|---|---|---|
| Thermocycling Conditions | ||||
| Step | Stage | Temperature | Time | Cycles |
| 1 | Initial denaturation | 95˚C | 3 minutes | 1x |
| 2 | Denaturation | 98˚C | 15 seconds | 14,16,18x |
| Annealing | 67˚C | 20 seconds | ||
| Elongation | 68˚C | 4 minutes | ||
| 3 | Final elongation | 72˚C | 5 minutes | 1x |
| 4 | Hold | 4˚C | ∞ | Hold |
Use Ampure XP 2x beads for DNA clean-up. It is recommended to complete final elution in 20µL- 30µLof distilled nuclease free water. CRITICAL STEP : Over-drying of Ampure beads before elution step will result in loss of material.
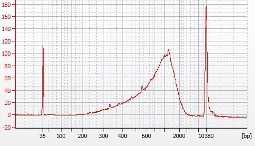
Measure DNA concentration using Qubit dsDNA kit. Based on Qubit results, take aliquots of sample and resuspend them to 1 ng/µL final concentration. Run these aliquots on a Bioanalyzer chip to confirm the size of WTA product. *Troubleshooting
A good WTA product trace from Bioanalyzer looks like so:
DAY 3: BEAD REINJECTION
Prepare a 2x PCR master mix.
Mix detection PCR reagents and beads in a 15mL falcon tube so that the final concentrations are:
| A | B | C | D |
|---|---|---|---|
| Reagent | Reagent conc. | Final conc. | Vol µL |
| TaqMan Assay (900 nM primers, 250 nM probe) | 20 X | 1 X | 50 |
| TaqPATH 2x Master Mix | 2 X | 1 X | 500 |
| Tween-20 | 10% (v/v) | 2.5% (v/v) | 50 |
| PEG-6000 | 20% (w/v) | 2.5% (w/v) | 50 |
| Beads | 350 | ||
| Total | 1 mL |
Soak for 1h 0m 0s on shaker in the dark at room temperature.
Spin 15ml tubes at 4500x g to separate PCR mix and beads. Separate beads and PCR mix.
Add PCR mix (20% of bead volume) to beads, vortex briefly. This prevents beads from aggregating in the device and makes reinjection stable.
Load syringes
- Load PCR mix into a 3 mL syringe with a HFE oil backing (no fluorosurfactant)
- Load beads into a 3 mL syringe
- Load Droplet generation oil for Evagreen into a 3 mL syringe
Place PCR mix, beads, and oil into the syringe pumps and connect the tubing to the reinjection device.
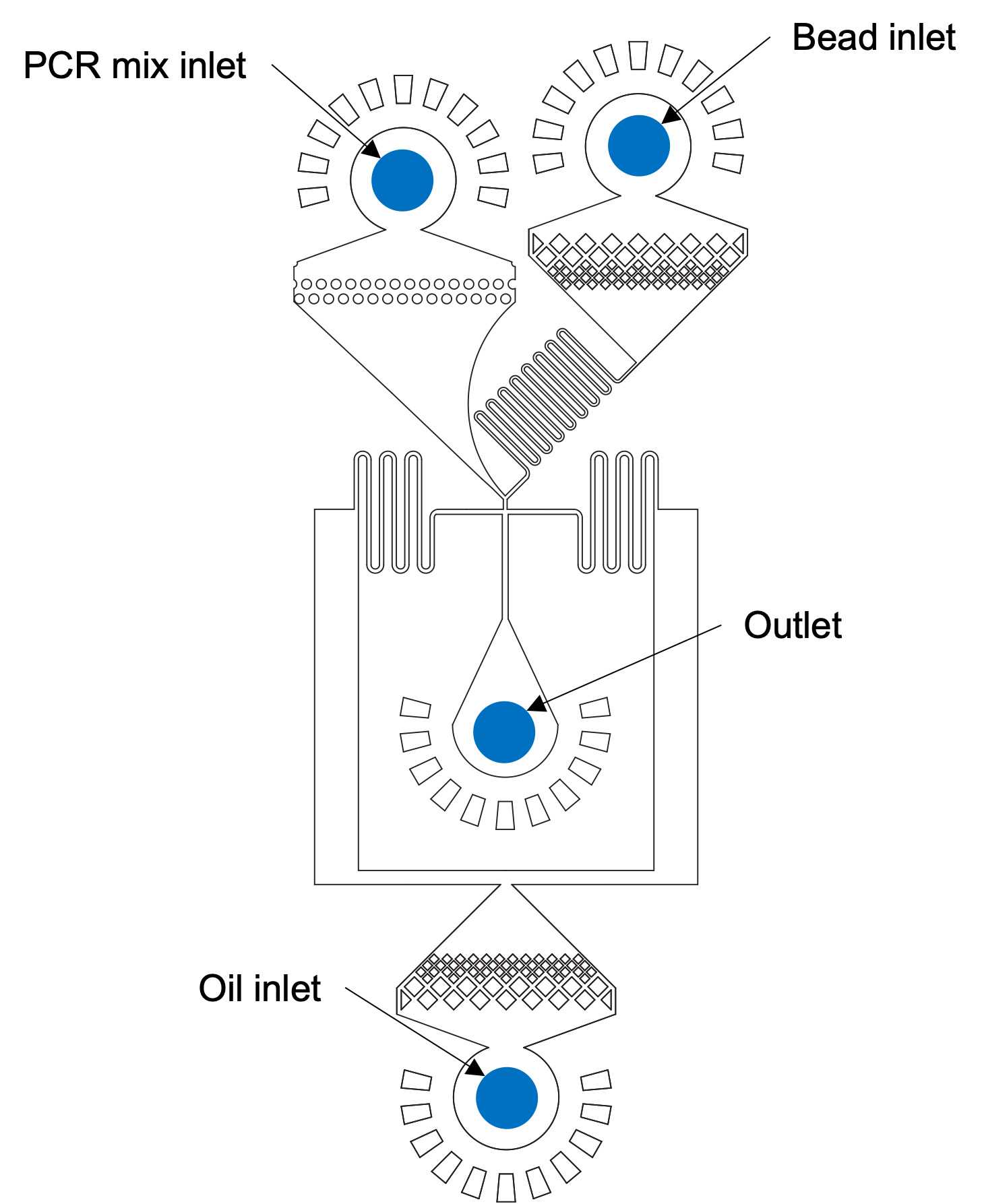
Start the reinjection device with the following flow rates, to create ~70 µm diameter droplets:
| A | B |
|---|---|
| Channel | Flow rate |
| PCR mix | 600 µL/hr |
| Beads | 400 µL/hr |
| Evagreen oil | 1800 µL/hr |
Begin reinjection, collecting 30µL aliquots into PCR strips. Occasionally during reinjection, verify that bead loading into droplets is close to 100% (or at least above 70%) by capturing videos.
After collection, thermocycle strips as follows with ramp rate set to 1.0 C/s
| A | B | C | D |
|---|---|---|---|
| Thermocycling Conditions | |||
| Stage | Temperature | Time | Cycles |
| 1 | 88˚C | 10 minutes | 1x |
| 2 | 88˚C | 30 seconds | 55x |
| 60˚C | 1 minute | ||
| 3 | 4˚C | ∞ | Hold |
DAY 4: SORTING DROPLETS
See previously published paper of Mazutis et. al (Nat. Protoc. 2013 May;8(5):870-91.) for details on droplet sorting procedure.
Load syringes
- Load thermocycled emulsion into a 3 mL syringe
- Load Droplet generation oil for Evagreen into a 3 mL syringes (Oil 1)
- Load HFE-7500 into a 3 mL syringes (Oil 2)
- Load HFE-7500 into a 3 mL syringes (Oil 3)
Place emulsion, and oil into the syringe pumps and connect the tubing to the sorting device. Fill the saltwater electrode and moat channels with 2 M NaCl solution. Connect saltwater electrode to voltage amplifier.
Flow rates for reinjecting into a detection/sorting device:
| A | B |
|---|---|
| Channel | Flow rate |
| Emulsion | 100 µL/hr |
| Spacer oil | 400 µL/hr |
| Oil 1 | 2000 µL/hr |
| Oil 2 | 3000 µL/hr |
| Air | 2-5 psi |
Determining cycle number for Amplification of sorted material
Cycling number is determined by performing WTA on a known number of sorted drops and calculating the number of cycles needed to obtain a desirable yield for positive drop sorts.
Sort 6-9 aliquots of the number of cells (# cells) that you will be collecting in the real experiment.
Perform WTA and bioanalyzer using 3 different cycle numbers in duplicate (or triplicate)
Plot the data and select the number of cycles that gives at least 2 ng/µL in 20µL elution volume, or 40ng of DNA, determined by qubit or bioanalyzer.
For Single drop sorts
Collect single drops into PCR strips.
After collection, overlay each tube with 37µL of water.
Spin tubes at 20000rpm.
Freeze tubes at -80°C for at least 2h 0m 0s or overnight. PAUSE POINT
Take tubes out of freezer and heat tubes to 60°Cfor 0h 10m 0s.
Using the table below, prepare a Master mix.
| A | B | C |
|---|---|---|
| Reagent | Reagent conc. | Vol. (for single tube) |
| KAPA buffer | 5 X | 10 µL |
| dNTP | 10 mM | 1.5 µL |
| PCR primer | 100 µM | 0.2 µL |
| Kapa polymerase | 1 µL | |
| Nuclease free water | - | 0.3 µL |
| Total | 13 µL |
Add 13µL of PCR reagents for 50µL total reaction volume.
Flick the tube to mix, spin, and thermocycle.
Thermocycle according to previously determined cycle number (Step 53).
Cleanup WTA with 1.2X Ampure XP.
Add 60µL Ampure XP beads to 50µL WTA reaction.
For 100 drop sorts
Collect drops in Eppendorf tubes.
Remove oil from Eppendorf tubes, leaving only a small amount to ensure that no unbroken drops are removed.
Cleanup WTA with 1.2X Ampure.
Add 80.4µL Ampure XP beads to 67µL WTA reaction.
Add a 50µL aqueous overlay (distilled nuclease-free water).
Spin tubes at 2000rpm. Then freeze tubes at -80°C for at least 2h 0m 0s or overnight. PAUSE POINT
Take tubes out of freezer. Heat tubes to 60°C for 0h 10m 0s. Remove samples from 60°C and carefully mix only the aqueous layer by pipet.
Carefully transfer the aqueous layer in tubes to PCR strips. It is better to transfer some oil than to not transfer all aqueous.
Set up the PCR master mix as follows:
| A | B | C |
|---|---|---|
| Reagent | Reagent conc. | Vol. (for single tube) |
| KAPA buffer | 5 X | 10 µL |
| dNTP | 10 mM | 1.5 µL |
| PCR primer | 100 µM | 0.2 µL |
| Kapa polymerase | 1 µL | |
| Nuclease free water | 4.3 µL | |
| Total | 17 µL |
Add 17µL of PCR master mix to 50µL of aqueous layer
Carefully mix by flicking the PCR tubes. Do not form emulsions. After mixing, spin tubes to separate aqueous and oil layers.
Thermocycle according to previously determined cycle number (Step 53).
Troubleshooting
| A | B | C | D |
|---|---|---|---|
| Step | Problem | Possible Reason | Solution |
| 21 | Cells are all dead or counts seem incorrect. | Automated Cell counter may not be able to differentiate between live and dead cells for smaller cell types like PBMCs. Cells may not have been resuspended thoroughly. | Mix cell resuspension thoroughly and count cells using a hemocytometer slide. |
| 29 | 1) Solution seems to have solidified. 2) Beads are not separating from hexane. | 1) Issue with agarose concentration. 2) Generated double emulsions during addition of HFE/PFO in step 27 | Restart Experiment |
| 34 | 1) Beads not passing through the filter. 2) Film of beads on filter leading to large loss | 1) Beads are larger than normal (> 55 µm) and struggle to pass through 100 µm filter. 2) Beads are too concentrated. | Split original tube from which beads came from into multiple tubes. Use multiple filters if necessary, adding Wash 2 Buffer to help correctly sized beads pass through the filter. |
| 53 | Yield of DNA material obtained after WTA reaction is low/zero | Low/zero yield may be due to failure of reverse transcription reaction or the WTA PCR. Low/zero yield may be due to mRNA loss during initial washes before reverse transcriptase reaction. This happens when buffers used are not high enough salt concentration or when beads are not kept on ice and mRNA dissociates from the poly-T tail within conjugated agarose beads. | Restart Experiment |
