Cost-conscious generation of multiplexed short-read DNA libraries for whole-genome sequencing
Scott Ferguson, Ashley Jones, Justin Borevitz, Benjamin Schwessinger, David Stanley, Norman Warthmann
Abstract
Massively parallel, second-generation short-read DNA sequencing has become an integral tool in biology for genomic studies. Offering highly accurate base-pair resolution at the most competitive price, the technology has become widespread. However, high-throughput generation of multiplexed DNA libraries can be costly and cumbersome. Here, we present a cost-conscious protocol for generating multiplexed short-read DNA libraries using a bead-linked transposome from Illumina. By preparing libraries in high-throughput with small reaction volumes that use 1/50th the amount of transposome compared to Illumina DNA Prep tagmentation protocols, the cost per library can be substantially reduced, by approximately 1/20th. Furthermore, we optimised the protocol to minimise magnetic bead-based clean-ups between steps, further reducing cost, time and DNA input requirements. By developing our own dual index primers to multiplex nine 96-well microplates, up to 864 samples can be placed on a single flow cell. This enables efficient usage of large-scale sequencing platforms, such as the Illumina NovaSeq 6000, which offers up to three terabases of sequencing per S4 flow cell.
Before start
In this protocol, the transposome being used will be diluted 1/50, therefore up to 50 times more tagmentation reaction buffer is required. Prepare the following custom tagmentation buffer in adavance (example ordering details are in the materials section).
- 2x TMP: 20 mM Tris-HCl pH 8, 10 mM MgCl2, 16% PEG 8,000.
- Final concentration in tagmentation reaction is 10 mM Tris-HCl, 5 mM MgCl2, 8% PEG 8,000.
- Filter sterilise the solution and store at 4°C (fridge) for up to one year.
| A | B | C | D | E |
|---|---|---|---|---|
| Reagent | Target concentration | MW | Stock concentration | From stock |
| Tris-HCl (pH 8) | 20 mM | 121.14 | 1 M | 20 µL |
| MgCl2 | 10 mM | 95.21 | 1 M | 10 µL |
| PEG 8,000 | 16% | 8,000 | 25% | 640 µL |
| Nuclease-free water | NA | NA | NA | 330 µL |
| - | - | - | - | Total: 1 mL |
Steps
DNA quantification
Quantify the DNA samples. For a low-throughput number of samples, quantify with a Qubit Fluorometer according to the manufacturer's instructions (Thermo Fisher Scientific). The following describes quantification in high-throughput (96-well microplates) by reading on a fluorescent microplate reader.
To measure a 96-well microplate of DNA samples with a dsDNA quantification high sensitivity kit (for microplate reader) (BioDynami or Thermo Fisher Scientific), ensure concentrations are ≤ 33 ng/µL. For instance, first quantify a few samples on a Qubit Fluorometer. Highly concentrated microplates of DNA can be diluted 1/10 before starting (e.g. 5 µL of DNA added to 45 µL nuclease-free water in a new 96-well microplate).
Create a quantification working solution by mixing dsDNA HS dye/reagent 1:200 with dsDNA buffer. Each reaction will require 97 µL of the working solution and 3 µL of DNA sample (or standard) will be added later (100 µL total volume). For a whole 96-well microplate, create enough buffer for 96 samples, 32 standards and 22 extra samples dead volume as follows:
| A | B | C | D | E | F | G |
|---|---|---|---|---|---|---|
| Number of plates | Samples | Standards | Dead volume | Total | dsDNA buffer | dsDNA HS reagent |
| 1 | 96 | 32 | 22 | 150 | 14,477 µL | 73 µL |
| 2 | 192 | 64 | 44 | 300 | 28,954 µL | 146 µL |
For each 96-well microplate of DNA samples, prepare two microplates suitable for a fluorescent microplate reader. As standards will need to be read in addition to samples, the orginal 96-well microplate of samples will need to be split across two microplates (label as microplate A and B, or 1-48 and 49-96 etc).
Transfer 97 µL of quantification working solution into the first two columns of each microplate (16 wells each microplate). This will be used for 8 standards, done in duplicate on each microplate.
Transfer 97 µL of quantification working solution into the microplates for half of the the DNA samples. For example, filling columns 4-9 on microplate A, and 5-10 on microplate B, as follows:
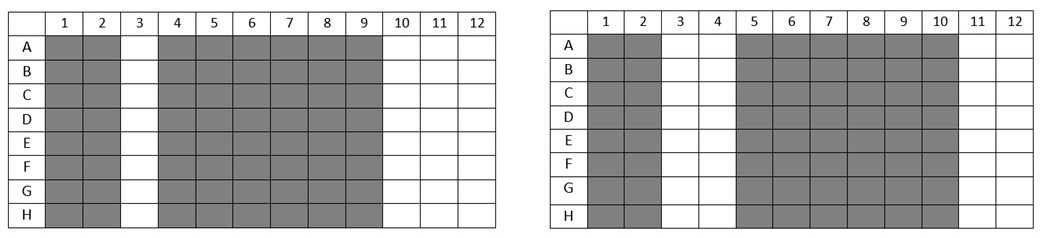
Add 3 µL of each standard to the microplate columns dedicated to standards. Perform this in duplicate.
Add 3 µL of each DNA sample to the microplate columns dedicated to samples.
Seal the microplate with adhesive film, vortex and briefly spin down. Incubate for 5 min.
Measure fluorescence using a microplate reader, using standard fluorescein wavelengths, for example excitation/emission at ∼480/530 nm.
Utilising the known concentration of standards included on the microplate, create a standard curve to determine DNA concentration of the other samples.
DNA dilution
Dilute all DNA samples to 1-2 ng/µL in a volume of approximately 25 µL. For example, aim for 1.5 ng/µL for each sample. First transfer nuclease-free water to a new 96-well PCR microplate and then add the appropriate amount of DNA.
Ensure the diluted samples are mixed and briefly spin down if necessary. Can be sealed with adhesive film and stored at -20°C until ready for tagmentation.
If necessary, the DNA can be quantified and diluted again as previously described.
Tagmentation
Pre-heat a PCR machine or heat block at 53°C suitable for a 384-well microplate. Will be used for the tagmentation reaction later.
Resuspend the Bead-Linked Transposome (BLT), thoroughly by flick-mixing and brief vortexing.
Prepare a tagmentation master mix in a 1.5 mL microcentrifuge tube. For an entire 96-well microplate of samples, prepare for 102 samples (i.e. a dead volume of 6 samples), as follows.
| A | B | C |
|---|---|---|
| Reagent | Per sample | For 102 samples |
| 2x TMP (custom buffer) | 3 µL | 306 µL |
| Transposome (BLT) | 0.2 µL | 20.4 µL |
| DNA (at 1-2 ng/µL) | 2.8 µL | - |
| TOTAL | 6 µL | 326.4 µL |
| Aliquot per sample | - | 3.2 µL |
Transfer 3.2 µL of the tagmentation master mix into 96-wells of a 384-well microplate (for example, the first quadrant in each set of four wells).
Transfer 2.8 µL of DNA (at 1-2 ng/µL) to each of the 96-wells containing tagmentation master mix. Gently pipette mix after the transfer.
Seal the microplate with adhesive film (PCR-grade), lightly vortex, keeping the liquid in the bottom of the well to avoid centrifuging.
Incubate microplate at 53°C for 30 min, mixing at 300-450 rpm if possible. If not in a shaker, mix by lightly vortexing after 15 min then put back for another 15 min.
After incubation, remove microplate from heat block and keep at room temperature until PCR.
PCR for enrichment and barcoding
Prepare a PCR master mix without the index primers and the tagmentation reaction (the template). For an entire 96-well microplate of samples, prepare for 102 samples (i.e. a dead volume of 6 samples). Alternatively, if using an automated workstation e.g. JANUS G3 (PerkinElmer), prepare for 28 samples in quadruplicate (i.e. 24 samples and 4 extra as dead volume), in four separate 1.5 mL microcentrifuge tubes. Prepare as follows.
| A | B | C | D |
|---|---|---|---|
| Reagent | Per sample | For 102 | (For 28) x4 |
| Nuclease-free water | 28.15 µL | 2,871.3 µL | 788.2 µL |
| 5x Q5 reaction buffer | 7.5 µL | 765 µL | 210 µL |
| 10 mM dNTPs | 1 µL | 102 µL | 28 µL |
| Q5 High-Fidelity DNA Polymerase | 0.35 µL | 35.7 µL | 9.8 µL |
| 2.5 µM i5 index primer | 4 µL | - | - |
| 2.5 µM i7 index primer | 4 µL | - | - |
| Tagmentation reaction | 5 µL | - | - |
| TOTAL | 50 µL | 3,774 µL | 1,036 µL |
| Aliquot per sample | - | 37 µL | 37 µL |
Transfer 37 µL of PCR master mix to each well of a 96-well PCR microplate.
Add 4 µL of a 2.5 µM i5 index primer and 4 µL of a 2.5 µM i7 index primer to each sample. Ensure each well has a unique combination of dual index primers.
Transfer 5 µL of tagmentation reaction (template) from the 384-well microplate to the 96-well microplate containing the PCR master mix aliquots.
Seal microplate with adhesive film (PCR-grade), vortex and briefly centrifuge (avoid pelleting the beads).
Perform PCR as follows:
72°C for 3 min
98°C for 3 min
12-16 cycles of:
- 98°C for 45 s
- 62°C for 30 s
- 72°C for 2 min
72°C for 1 min
Hold at ≤ 10°C
Proceed to pooling or store at -20°C.
Pool libraries
Quantify the DNA libraries as previously described, using a dsDNA quantification high sensitivity kit for microplate reader (BioDynami or Thermo Fisher Scientific), or a Qubit Fluorometer for low-throughput samples.
Add 40 µL of nuclease-free water into 1.5 mL microcentrifuge tube to start the pool.
Transfer an equal amount of each library (based on concentration, molarity or use a set volume e.g. 10 µL) from each well of the microplate into the same 1.5 mL microcentrifuge tube. This can be performed on an automated workstation (e.g. JANUS G3 by PerkinElmer), providing a csv file with volumes to transfer for each sample.
Place the tube on a magnetic rack for 5 min, or until all beads pellet.
Keeping the tube on the magnetic rack, transfer the supernatant to a new tube, without disturbing the beads. The beads can be discarded.
Clean-up and size selection (AMPure XP)
The pooled library can now undergo clean-up and size selection, which is flexible and multiple methods exist. For example, use AMPure XP Reagent (SPRI paramagnetic beads) according to the manufacturer's instructions (Beckman Coulter). Typically, 0.5-1x of the beads are addded relative to the total volume of the pool.
It may be necessary to repeat the clean-up to remove all primers and short fragments from the pool. For example a second 0.6x AMPure XP size selection according to the manufacturer's inctructions (Beckman Coulter).
Alternatively (or additionally), the library can udergo clean-up and size selection using a PippinHT (Sage Science), as detailed below.
Clean-up and size selection (PippinHT)
Prior to PippinHT size selection, the library may need to be concentrated, which can be done with AMPure XP beads (or equivalent) according to the manufacturer's inctructions (Beckman Coulter).
To perform a stringent size selection, a PippinHT (Sage Science) can be used. This will remove undesirable shorter fragments that will have overlapping sequencing on paired-end platforms. Using a PippinHT, perform an automatic size selection and gel purification, following the manufacturer’s instructions (Sage Science). A 2% agarose casette and reagents are ideal for Illumina short-read libraries. A size selection of 430 - 600 bp is ideal for Illumina 150 bp paired-end sequencing, but will depend on sequencing platform and the quantity of pooled libary required.
After PippinHT size selection, the library can be cleaned and concentrated with AMPure XP beads (or equivalent) according to the manufacturer's inctructions (Beckman Coulter).
Preparation for sequencing
Perform a quality check analysis of the final library pool on a high sensitive automated electrophoresis system, such as LabChip GX devices (PerkinElmer) or Bioanalyzer/TapeStation devices (Agilent Technologies).
Ensure the library looks as expected, relative to size selection and sequencing strategy. For Illumina NovaSeq 6000 150 bp paired-end sequencing, a library 400-600 bp is ideal. Estimate the average fragment length.
Quantify the library at least twice with a Qubit Fluorometer, using a Qubit dsDNA high sensitivity assay kit. Determine the average of the readings.
Calculate molarity; nM = (ng/µL x 106) / (660 g/mol x average fragment size bp).
Dilute the library to a suitable molarity for sequencing. Store at -20°C until ready for sequencing.
Send for sequencing, on ice, cold packs or dry ice depending on the distance to the closest sequencing facility.
