A modified method to analyse cell proliferation using EdU labelling in large insect brains
Amaia Alcalde Anton, Max S Farnworth, Laura Hebberecht, Jill Harrison, Stephen H Montgomery
Disclaimer
This protocol has been modified from the instructions of the Click-iT™ EdU Cell Proliferation Kit for Imaging, Alexa Fluor™ 488 dye - Thermofisher. Note that some steps (e.g. prepare the stock solutions) have not been changed.
Abstract
The study of neurogenesis is critical to understanding of the evolution of nervous systems. Within invertebrates, this process has been extensively studied in Drosophila melanogaster , which is the predominant model thanks to the availability of advanced genetic tools. However, insect nervous systems are extremely diverse, and by studying a range of taxa we can gain additional information about how nervous systems and their development evolve. However, studying this variation requires adapting labelling techniques to visualise cell division in less commonly studied organisms, as methods developed for common laboratory insects often do not work. Here, we present a modified protocol for EdU staining to examine neurogenesis in large-brained insects, using Heliconiini butterflies as our primary case, but also demonstrating applicability to cockroaches, another large-brained insect.
Steps
0. BEFORE YOU START – PREPARE THE STOCK SOLUTIONS
Prepare a 10 mM stock solution of EdU: Add 2mL of DMSO to EdU, then mix well (store at ≤20°C).
Prepare a working solution of the Alexa Fluor® azide: Add 70µL of DMSO to the Alexa Fluor® azide (store at ≤20°C).
Prepare a working solution of 1X Click-iT® EdU reaction buffer: Transfer all of the solution (4 mL) in the 1X Click-iT® EdU reaction buffer bottle to 36mLof deionized water. Store at4°C.
To create the 10X stock solution of the Click-iT® EdU buffer additive, add 2mL of deionized water to the vial (store at ≤ 20°C).
Prepare paraformaldehyde, PFA (4% PFA in [0.1 M] phosphate-buffered saline (PBS; 7.4 pH). Alternatively, zinc-formaldehyde, ZnFA (0.25% [18.4 mM] ZnCl2; 0.788% [135 mM] NaCl; 1.2% [35 mM] sucrose; 1% formaldehyde) can also be prepared and used. Store at 4Room temperature.
Prepare the permeabilization buffer (1 % Triton ® X-100 in [0.1 M] PBS). Store at 4°C.
1. EDU INCORPORATION
Prepare the EdU working solution. Dilute EdU in Grace’s Medium (GM, ThermoFisher, #11595030) to a final concentration of 20µM: add 20µL from the stock solution (prepared in step 0.1) to 10mL of GM. Make 1 mL aliquots (store at ≤ 20°C).
A) Ex vivo brain incubation
Dissect out the brain in 50% Grace’s medium diluted in [0.1 M] phosphate-buffered saline (PBS; pH
7.4).
Incubate the brain in a solution of EdU diluted in Grace’s medium for 1-2h.
B) In vivo In vivo incubation
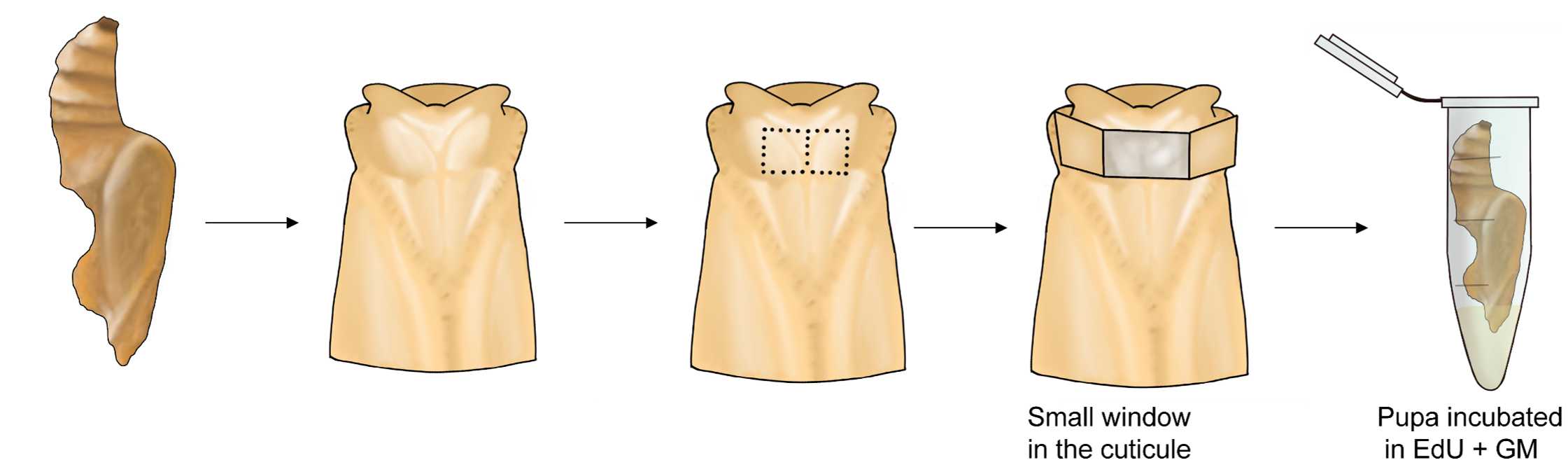
For active life stages, chill individuals in a refrigerator for a couple of minutes to anesthetize them. When ready, open a small window in the cuticle.
Incubate the brain in a solution of EdU diluted in Grace’s medium for 1-2h.
After the incubation, finish the dissection in HEPES-buffered saline, HBS ([150 mM] NaCl;
[5 mM] KCl; [5 mM] CaCl2; [25 mM] sucrose; [10 mM HEPES]; pH 7.4).
2. FIXATION
Transfer the brains to a 12 well plate, with up to 5 brains per well.
Fix the brain in a solution of paraformaldehyde, PFA (4% PFA in [0.1 M] phosphate-buffered saline (PBS; 7.4 pH) for 6-14h (Depending on the stage 14h for adults, 6-10h for larval stages).
Remove the fixative and wash each well three times for 10 min with 1mL of 0.1% Triton® X-100 in [0.1 M] PBS, 0.1%PBS-T.
3. PERMEABILIZATION
Remove the PBS solution (from step 12).
Add 1mL of 1% Triton® X-100 in [0.1 M] PBS, 1%PBS-T to each well.
Incubate at room temperature for 2h for whole brains, or 30 minutes for
sectioned tissue.
4. CLICK IT REACTION
Dilute 1X Click-iT® EdU buffer additive in deionized water at a ratio of 1:10. This should be prepared fresh and used on the same day.
Remove the permeabilization buffer (from step 3.15).
Wash each well three times with 0.1% Triton® X-100 in [0.1 M] PBS, 0.1%PBS-T.
Remove the wash solution.
Prepare the Click-IT® reaction mix. Add the ingredients in the order defined by the manufacturer, replicated below. Use the Click-iT® reaction mix within 15 minutes of preparation.
Table 1. Click-iT® reaction cocktail. All the components should be added in the order listed below. Table from Click-iT® EdU Imaging Kits Protocol, ThermoFisher.
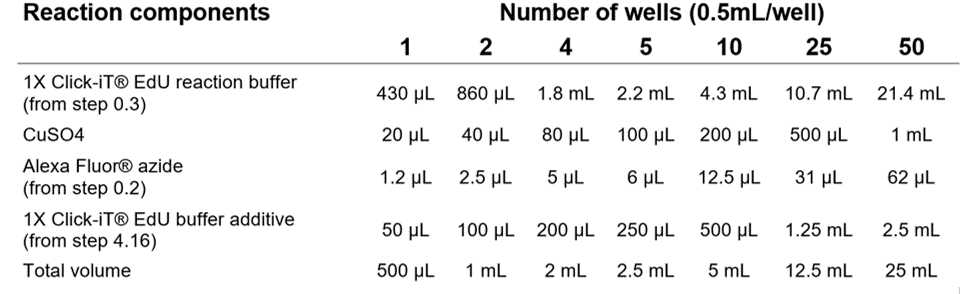
Add 0.5mL of the Click-iT® reaction cocktail to each well containing up to 3 brains, and 1mLto each well containing 4-5 brains. Make sure the brains are completely covered in the reaction mix.
Incubate the plate for 30 minutes at room temperature, protect from light using aluminium foil.
Remove the reaction mix.
Wash each well three times with 1mL of 0.1% PBS-T (10 minutes each).
Remove the wash solution.
NUCLEAR STAINING with Hoechst 33342 or DAPI Staining
A) Hoechst 33342
Wash each well twice with 1mL of [0.1 M] PBS (30 minutes each).
Remove the wash solution.
Dilute the Hoechst 33342 solution in [0.1 M] PBS at a ratio of 1:2000 to obtain a 1X Hoechst 33342 solution (the final concentration is 5 µg/mL).
Add 1mL of 1X Hoechst 33342 solution per well. Incubate for 2-3 hours for whole brains, or 30 minutes for sectioned tissue, at room temperature, protected from light .
Wash each well twice with 1mL of PBS.
Remove the wash solution.
B) DAPI
Wash each well twice with 1 mL of [0.1 M] PBS (30 minutes each).
Remove the wash solution
Wash once in milliQ/deionized H2O with 0.2% Triton, 10 minutes.
Dilute the DAPI solution in miliQ H2O at a ratio of 1:1000.
Add 1 mL of diluted DAPI solution per well. Incubate for 2-3 hours for whole brains, or 30 minutes for sectioned tissue, at room temperature, protected from light .
Remove the DAPI solution.
Wash each well with 1 mL of miliQ H2O with 0.2% Triton for 10 minutes.
Wash each well three times with 1mL of 0.1% PBS-T.
6. TISSUE CLARIFICATION
Incubate the brains under agitation in progressively more concentrated glycerol solutions: 1%, 2%, 4% (2 h each), 8%, 15%, 30%, 50%, 60%, 70%, and 80% (1 hour each) glycerol diluted in 0.1M Tris buffer, with DMSO to 1% final concentration.
Wash the brains with 100% ethanol for 30 minutes under agitation, repeat three times.
Transfer each brain in a small amount of ethanol (200-400µL) to an Eppendorf tube.
Underlay the ethanol with methyl salicylate, wait for brain to sink with no agitation.
Aspirate the fluid from the top down, replace with fresh methyl salicylate and allow the brain to sink again for ~30 minutes with no agitation .
Aspirate the fluid and mount in fresh methyl salicylate.
