snm3C-seq3
Chongyuan Luo, Hanqing Liu, Anna Bartlett, Rosa Castanon, Yi Zhang, Joseph R. Ecker
Abstract
Steps
Prepare Single Nuclei
Prepare stock solutions, stored at 4°C
1. NIM: Sucrose (250 mM), KCl (25 mM), MgCl2 (5 mM), Tris-Cl pH 8.0 (10 mM)
2. Dilutent: Tris-Cl pH 8.0 (120 mM), KCl (150 mM), MgCl2 (30 mM)
Prepare the following solutions fresh before each experiment
-
NIMT (on ice): NIM + Triton X-100 (0.1%) + DTT (1 mM) + Proteinase Inhibitor (1:100
dilution) -
50% Iodixanol (Room temp) : 5 vol. Optiprep (60% Iodixanol) + 1 vol Dilutent
-
25% Iodixanol (Room temp) : 1 vol. 50% Iodixanol + 1 vol NIM
-
DPBS + 1% BSA (on ice)
Prepare nuclei:
-
Fast cool centrifuge with swinging bucket tube rotor to 4°C.
-
Remove frozen tissue from -80°C freezer, place on ice.
-
Transfer tissue with 2.4 mL NIMT from tube into the large dounce.
-
Use loose pestle A 40 times gently without introducing bubbles.
-
Use tight pestle B 40 times gently without introducing bubbles.
-
Transfer lysed solution into a 5-mL or 15-mL tube and keep on ice.
-
If doing NeuN stain: Add 6 μL of NeuN 488 (1:500 dil). Mix and incubate 15 min on ice, keeping solution covered from light.
-
Mix the lysis suspension with 1.5 mL of 50% Iodixanol by pipetting.
-
Slowly pipette 1 mL of cell mixture onto 500 μL 25% Iodixanol cushion, 4 x 2-mL tubes in total per sample.
-
Centrifuge in swing rotor for 20 min at 10,000 x g at 4°C.
Nuclei staining and count:
-
Remove supernatant very carefully, avoiding debris.
-
Using ice-cold DPBS+RNase Inhibitors, resuspend and combine the pellets from each tube into a total of 1 mL nuclei solution in a new tube.
-
Add Hoechst 33342 (dil: 1:1000): Dilute the Hoechst 1:10 (5 µL + 45 µL DPBS), then add 5 µL to 1 mL sample.
-
Incubate on ice for 5 min.
-
Mix 10 µL Nuclei suspension with 10 µL Trypan Blue and mix by pipetting.
-
Add 10 µL stained solution to a Cell Counter Slide and read using the Bio-Rad Cell Counter TC-20.
Note: Nuclei count is Total Count minus Live Count
3C Protocol
Crosslinking:
-
Split sample into 2 tubes (1-2 million nuclei in each tube) and bring volume up to 1 mL with DPBS.
-
Add 57 µL of 37% formaldehyde (for a final concentration of 2%) to each tube and mix well by inverting 10 times.
-
Incubate 10 min (5 min if a NeuN pre-stain was done to avoid stain degradation) at room temperature, inverting occasionally.
-
Add 91.9 µL Stop Solution (or 2.5 M Glycine) and mix well by inverting 10 times.
-
Incubate for 5 min at room temperature, inverting occasionally.
-
Place sample on ice and incubate an additional 5 min.
-
Pellet cells by centrifuging 5 min at 2500 x g or 10 min at 1000 x g.
-
Discard supernatant, being sure to leave no residual liquid, and resuspend in 1 mL of 1x PBS.
-
Repeat steps 7-8 two more times to completely wash away residual formaldehyde/glycine.
-
Proceed directly to next section or freeze samples at -80°C up to 5 days.
3C Nuclei Conditioning:
-
Resuspend each reaction of crosslinked nuclei in 20 µL DPBS.
-
Add 24 µL Conditioning Solution and mix gently by pipetting.
-
Incubate at 62°C for 10 min. If using a thermal cycler, set the lid temperature to 85°C.
-
Add 20 µL Stop Solution 2 and mix gently by pipetting.
-
Incubate at 37°C for 15 min. If using a thermal cycler, set the lid temperature to 85°C.
3C Enzymatic Reactions
Note: Some of the next steps require addition of several reagents in the same step. These reagents should be combined into master mixes using the following tables (including 10% extra to accommodate distribution).
- Digestion: Add 28 µL of master mix containing the following reagents:
| A | B | C | D | E | F |
|---|---|---|---|---|---|
| Reagent | 1 Rxn (µL) | w/10% extra | # Rxns | Final Volume | |
| 1x CutSmart | 12 µL | 13.2 µL | x | ||
| 10x CutSmart (or Buffer H) | 7 µL | 7.7 µL | x | ||
| NLAIII/Enzyme H1 | 4.5 µL | 4.95 µL | x | ||
| MboI/Enzyme H2 | 4.5 µL | 4.95 µL | x | ||
| Total | 28 µL |
-
Mix gently by pipetting and incubate as follows. If using a thermal cycler, set the lid temperature to 85°C.
-
37°C for 60 min (or up to overnight)
-
65°C for 20 min
-
25°C for 5 min
-
-
Mix gently by inversion.
-
Ligation: Add 82 µL of master mix containing the following reagents:
| A | B | C | D | E | F |
|---|---|---|---|---|---|
| Reagent | 1 Rxn (µL) | w/10% extra | x | # Rxns | Final Volume |
| Buffer C | 70 µL | 77 µL | x | ||
| Enzyme C | 12 µL | 13.2 µL | x | ||
| Total | 82 µL |
-
Mix gently by pipetting and incubate at room temperature for 15 min.
-
Samples can be stored at 4°C overnight or continue to FACS.
FACS
Prepare samples for FACS:
-
Combine the reactions for each sample and add DPBS + 1% BSA up to a total volume of 1 mL.
-
Centrifuge at 1,000 x g for 10 min at 4°C in swinging bucket rotor.
-
Remove supernatant and gently resuspend in 200 µL DPBS + 1% BSA. Add another 800 µL DPBS + 1% BSA to bring total volume up to 1 mL.
-
Add 5 µL 1:10 diluted Hoecsht 33342.
-
Add 1 µL of antibody to enhance labeling.
-
Incubate on ice for 5 min.
-
Filter the cells through a 40 µm cell strainer and transfer into a polypropylene tube for sorting. Keep on ice until ready to sort.
Single nuclei sorting:
-
Thaw and spin down (5 sec at 1000 x g) 384-well plates containing 1 µL digestion buffer.
-
Sort single nuclei into wells using BD Influx or other sorter.
- Use 2N gating for Hoechst stain to exclude debris and nuclei in cell replication stages. See example gate with Hoechst on the x-axis and side scatter on the y-axis. Debris is seen to the left of the gate and possible replicating nuclei are to the right.
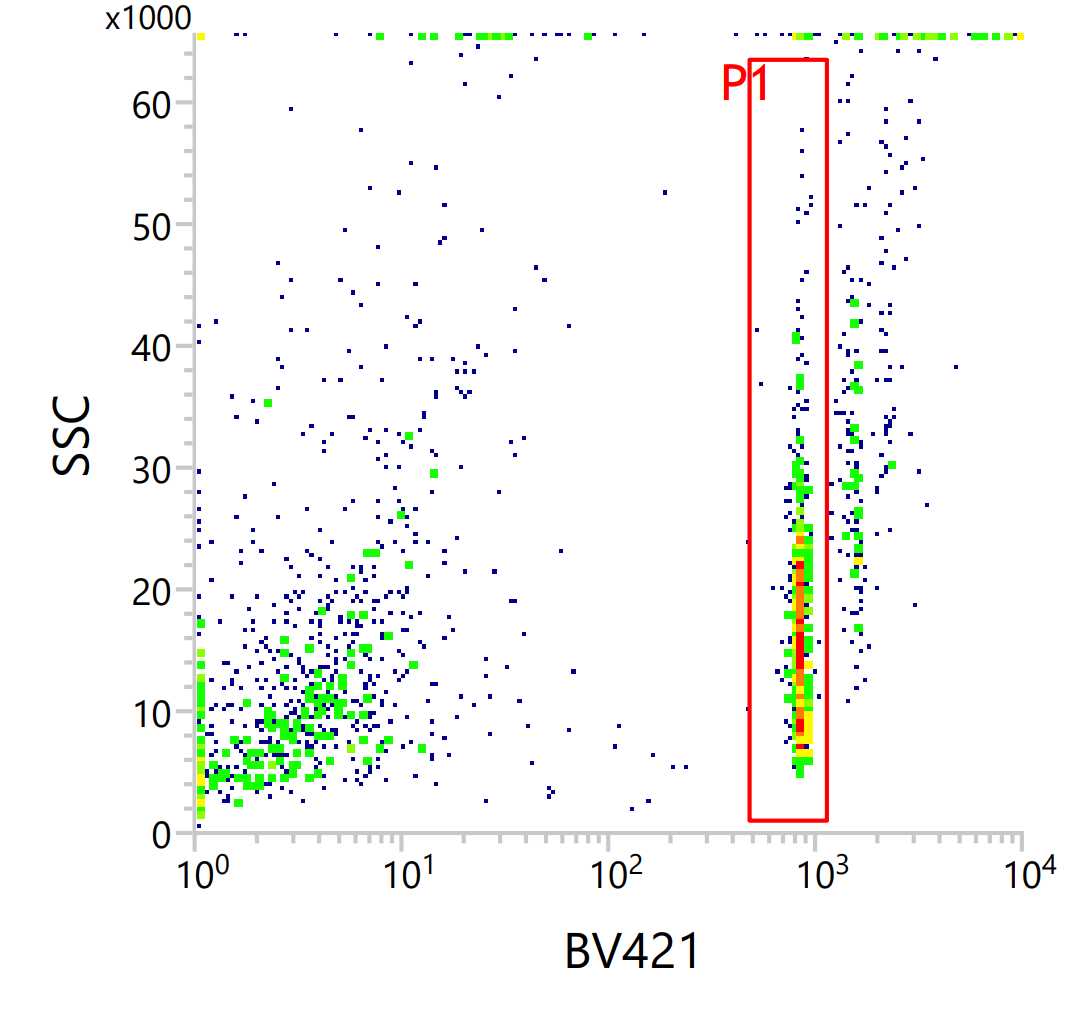
b. If NeuN 488 stained: Sort NeuN+ in col 1-22 and NeuN- in col 23-24
3. Sort using 1 drop single mode
3. After sorting spin down the sample plates for 5 sec at 1000 x g.
-
Incubate plates for 20 min at 50°C to run proteinase K reaction.
-
Plates can be stored at -20°C for up to 1 year.
Bisulfite Conversion
Prepare the Zymo CT Conversion Reagent (3 bottles for 16 x 384-well plates)
-
Add 7.9 mL Solubilization Buffer and 3 mL Dilution Buffer to each Conversion Reagent bottle and mix for 10 minutes.
-
Add 1.6 mL Reaction Buffer to each Conversion Reagent bottle and mix for an additional 5 minutes.
Bisulfite Reaction:
*This step can be automated using Biomek i7
-
Add 6 µL CT Conversion Reagent to each well.
-
Seal, vortex, and quick spin (500 rcf) plates.
-
Incubate plates using [DIRECT] method on PCR machines with 384-well plate block.
-
98 °C for 8 minutes
-
64 °C for 3.5 hours
-
Hold at 4 °C for up to 20 hours
-
Bisulfite Cleanup:
*Steps in this section are automated for up to 8 x 384-well plates using Biomek i7
Prepare MagBead master plate:
a. Invert Magbead J stock tube repeatedly to ensure homogeneity.
b. Using an 8-channel pipette, aliquot 58 μL Magbead J stock to each well of a 96-well plate (avoid bubbles and do not spin). Tap plate to remove bubbles.
c. Distribute 14 μL beads to each well of a clean 384-well plate.
d. Tap down 384-well plate and pipette out any bubbles from the bottom of the wells (do not spin).
e. Distribute 1.5 μL to each well of sample plates.
(Optional) If running 16 plates, repeat steps 12:1-12:9 for the second set of 8 plates.
Bind the free DNA to the Magbeads:
a. Add 20 µL M-Binding Buffer to each well and mix by pipetting.
b. Incubate at room temperature for 5 minutes.
c. Move the plates onto the magnets.
d. Remove supernatant and discard in the waste trough.
Wash the beads:
a. With the plates still on the magnets, add 20 μL M-Wash Buffer to all plates and incubate at room temperature for 30 seconds.
b. Remove supernatant and discard in the waste trough.
Add M-Desulphonation Buffer to the plates:
a. Add 15 μL M-Desulphonation Buffer to all plates.
b. Incubate at room temperature for 15 minutes.
c. Remove supernatant and discard in the waste trough.
Wash the beads:
a. Add 20 μL M-Wash Buffer to all plates and incubate at room temperature for 30 seconds.
b. Remove supernatant and discard in the waste trough.
c. Repeat wash one additional time.
(Optional) Do an extra supernatant removal to further empty out wells if desired
Let the plates dry:
a. Let plates sit for 5 minutes. Keep them on the magnets to avoid beads jumping due to static.
b. Check that all the wash buffer has evaporated before moving on.
Elute the DNA from the beads using the RP elution plate:
a. Add 5.5 μL appropriate Random Primer solution to all plates (note the barcodes).
b. Remove the plates from the magnets and seal well.
c. DO NOT VORTEX; instead, throw the sealed plates onto the bench 10-15 times, until beads appear to be mixed into elution buffer. Repeat for all plates.
d. Quick spin plates for 5s at 200xg
e. Incubate at room temperature for 5 minutes. Place plates back onto the magnets.
Transfer eluted DNA into clean 384-well plates:
a. Transfer 5 µL sample elution into a clean, labeled 384-well plate
b. Seal new sample plates and quick spin for 5s at 500xg.
Random Priming
Random primed DNA synthesis:
**Steps in this section are automated for up to 16 x 384-well plates using Biomek i7
- Prepare appropriate volume of Random Priming Master Mix (RP MM) in a 50-mL conical tube and invert to mix.
| A | B | C | D |
|---|---|---|---|
| Reagent | Volume per rxn | Vol for 8 plates | Vol for 16 plates |
| Nuclease-free H2O | 3.45 µL | 12420 µL | 24840 µL |
| Enzymatics Blue Buffer (10x) | 1.025 µL | 3690 µL | 7380 µL |
| dNTPs (10 mM each) | 0.50 µL | 1800 µL | 3600 µL |
| Klenow Exo- (50 U/µL) | 0.025 µL | 90 µL | 180 µL |
-
Denature 8 sample plates at 98 °C for 3 minutes in thermocycler. Remove plates from PCR machines and immediately place on ice to prevent rehybridization. Proceed once bottom of each plate is cool to the touch.
-
Add 5 μL RP MM to each sample well. Seal, vortex, and quick spin (5s at 500xg) sample plates.
-
Incubate plates in thermocycler with the following program:
-
4°C for 5 minutes
-
25°C for 5 minutes
-
37°C for 60 minutes
-
Hold at 4 °C
-
-
Repeat steps 2-4 for second set of 8 plates (if applicable).
Inactivation of free primers & dNTPs:
**Steps in this section are automated for up to 16 x 384-well plates using Biomek i7
- Prepare appropriate volume of Exo/rSAP Master Mix (E/S MM) in a 15-mL
conical tube and invert to mix.
| A | B | C | D |
|---|---|---|---|
| Reagent | Vol per rxn | Vol per 8 plates | Vol per 16 plates |
| Nuclease-free H2O | 1.15 µL | 4140 µL | 8280 µL |
| Enzymatics Blue Buffer (10x) | 0.2 µL | 720 µL | 1480 µL |
| Exonuclease I (20 U/µL) | 0.1 µL | 360 µL | 720 µL |
| rSAP (1 U/µL) | 0.05 µL | 180 µL | 360 µL |
-
Add 1.5 μL Exo/Sap Master Mix to each sample well. Seal, vortex, and quick spin sample plates (5s at 500xg).
-
Incubate plates in thermocycler with the following program:
-
37°C for 30 minutes
-
Hold at 4°C
-
-
Repeat steps 2-3 for second set of plates (if applicable).
-
Either continue to Reformat and Cleanup or store at -20°C overnight.
Reformat and Sample Cleanups
Compress 16 x 384-well plates to 8 x 96-well plates and add SeraMagbeads
**Steps in this section are automated for 16 x 384-well plates using Biomek i7. Note that the bead cleanups for each set of plates will overlap to increase timing efficiency.
Thaw (if applicable) and quick spin (5s at 500xg) sample plates.
Compress 384-well plates 1-8 into four new 96-well plates 1-4. 8 wells will be combined into one well. One 384-well plate will be compressed to half a 96-well plate:
a. Combine samples from each quadrant of the top half of 384-plate 1 (Rows A-H) and
dispense into the top half of a clean 96-well plate (Rows A-D).
Eg: A1,A2,B1,B2 of 384-well plate combine into A1 of the 96-well plate.
A3,A4,B3,B4 of 384-well plate combine into A2 of the 96-well plate.
b. Combine samples from each quadrant of the bottom half of 384-plate 1 (Rows I-P) and dispense into top half of the same 96-well plate (Rows A-D).
Eg: I1,I2,J1,J2 of 384-well plate combine into A1 of the 96-well plate along with the A/B row
samples.
c. Continue this pattern of pooling with 384-well plate 2 going into the bottom half of the same 96-well plate. Plates 3-4 will go into a second 96-well plate, 5-6 into a third 96-well plate, and Plates 7-8 into a fourth 96-well plate.
Bind the free DNA to the Sera-Mag beads in 96-well plates 1-4:
a. Thoroughly mix Sera-Mag beads by repeatedly inverting bottle and pour Sera-Mag beads to the brim of a low-profile 96-well reservoir.
b. Add 73.6 μL beads into sample plates and pipette up and down to mix.
c. Let plates incubate at room temperature for at least 5 minutes.
Compress 384-well plates 9-16 into four new 96-well plates 5-8:
a. Combine 384-well plates 9-16 following the same pattern of pooling in Step 15.2 above. Plates 9-10 will go into a fifth 96-well plate, 11-12 into a sixth 96-well plate, Plates 13-14 into a seventh 96-well plate, and Plates 15-16 into a eighth 96-well plate.
Bind the free DNA to the Sera-Mag beads in 96-well plates 5-8:
a. Thoroughly mix Sera-Mag beads by repeatedly inverting bottle and pour Sera-Mag beads to the brim of a low-profile 96-well reservoir.
b. Place plates 1-4 onto 96-well magnet and incubate for at least 5 minutes.
c. Add 73.6 μL beads to sample plates 5-8 and pipette up and down to mix. Incubate for at least 5 minutes.
Perform bead cleanups on 96-well plates 1-8
Carefully remove supernatant from plates 1-4 and discard in waste trough.
Wash beads in plates 1-4 with 80% EtOH:
a. Keeping plates on the magnets, add 180μL of 80% EtOH to plates 1-4 and incubate at room temperature for at least 30 seconds. Subsequently remove and discard EtOH in the waste trough.
b. Repeat for a second wash.
c. Remove plates from the magnets and incubate until beads are dry. The beads should appear cracked and there should be no visible droplets of EtOH in the wells.
Remove supernatant and add EtOH to 96 plates 5-8.
a. Carefully remove supernatant from plates 5-8.
b. Add 180 μL EtOH to plates 5-8 and incubate for at least 30 seconds.
Elute DNA from plates 1-4 and perform second EtOH wash on plates 5-8:
a. Once beads are dry, add 10 μL EB to plates 1-4. Set a 5-minute timer upon completion of transfer.
b. Seal plates, vortex until beads no longer remain on the sides of the well, and quick spin 5s @ 150xg to avoid pelleting beads. When 5-minute timer is completed, replace plates on the magnets.
c. Remove EtOH from plates 5-8 and discard in the waste trough.
d. Repeat EtOH wash of plates 5-8.
e. Remove plates from the magnets and incubate until beads are dry.
Elute DNA from plates 5-8:
a. Once plates are dry, add 10 μL EB to plates 5-8. Start a 5-minute timer upon completion of transfer.
b. Seal plates, vortex until beads no longer remain on the sides of the well, and quick spin 5s @ 150xg to avoid pelleting beads. When 5-minute timer is completed, replace plates on the magnets.
Compress 8 x 96-well plates to 1 x 96-well plate
Further compress plates 1-4 into a clean 96-well plate (Rows A-D). 4 wells will combine into 1 well of the new plate. Each original 384-well plate will be contained in one row of the 96-well plate.
a. Carefully unseal plates 1-4, still on the magnets.
b. Transfer eluted sample from plates 1-4 into a new 96-well plate.
c. Combine rows A-D of plate 1 into row A of the new plate
d. Combine rows E-H of plate 1 into row B of the new plate.
e. Continue this pattern with A-D of plate 2 in row C of the new plate, etc, until rows E-H of plate 4 are in row H of the new plate.
Further compress 96-well plates 5-8 into new 96-well plate. 8 wells will combine into 1 well of the new plate. Each original 384-well plate will be contained in one row of the 96-well plate.
a. Transfer eluted sample from plates 5-8 into a new 96-well plate.
b. Combine rows A-D of plate 5 into row A of the new plate
c. Combine rows E-H of plate 5 into row B of the new plate.
d. Continue this pattern with A-D of plate 6 in row C of the new plate, etc, until rows E-H of plate 8 are in row H of the new plate.
Compress the two 96-well plates into single new 96-well plate.
a. Transfer Columns 1-6 of first 96-well plate into Columns 1-6 of a new 96-well plate.
b. Transfer Columns 7-12 of first 96-well plate into Columns 1-6 of the new 96-well plate
to combine with previous transfer.
c. Transfer Columns 1-6 of second 96-well plate into Columns 7-12 of the new 96-well plate.
d. Transfer Columns 7-12 of second 96-well plate into Columns 7-12 of the new 96-well plate to combine with previous transfer.
Note: final plate configuration will have six wells per original 384-well plate i.e., wells A1-A6 contain plate 1, wells B1-B6 contain plate 2, etc. for the rest of the left half of the plate. Wells A7-A12 contain plate 9, wells B7-B12 contain plate 10, etc. for the rest of the right half of the plate. te.
Perform bead cleanup on compressed 96-well plate.
Bind free DNA to the Sera-Mag beads:
a. Thoroughly mix Sera-Mag beads by repeatedly inverting bottle and pour beads to the brim of the bead reservoir.
b. Add 64 μL beads to each well of the sample plate and pipette up and down to mix. Incubate
at room temperature for 5 minutes.
c. Move plate to plate magnet and wait until beads are settled to continue (approximately 3 minutes).
d. Carefully remove supernatant and discard in waste trough.
Wash beads with 80% EtOH:
a. Keeping plates on the magnets, add 180μL of 80% EtOH to plates 1-4 and incubate at room temperature for at least 30 seconds. Subsequently remove and discard EtOH in the waste trough.
b. Repeat for a second wash.
c. Remove plates from the magnets and incubate until beads are dry.
Elute DNA
a. Add 10 μL EB to each well of the sampleplate. Set a 5-minute timer.
b. Seal plate, vortex until beads no longer cling to sides of wells, then quick spin
for 5s at 100xg.
c. After 5-minute timer is complete, unseal plate and place on magnet.
d. Once beads have settled on the magnet (about 30 seconds), transfer elution to a
clean 96-well plate.
e. Check plate to ensure volume of wells appear even. Seal and quick spin (500xg).
DNA amplification
Adaptase addition
*Steps in this section are automated for 8 or 16 original 384-well plates using Biomek i7
- Prepare appropriate volume of Adaptase Mix in a 1.5 mL microcentrifuge tube and keep on ice.
| A | B | C | D |
|---|---|---|---|
| Reagent | Vol per rxn | Vol per 8 plates | Vol per 16 plates |
| Elution Buffer (EB) | 4.45 µL | 239.2 µL | 478.4 µL |
| Buffer G1 | 2.00 µL | 112.5 µL | 225.0 µL |
| Reagent G2 | 2.00 µL | 112.5 µL | 225.0 µL |
| Reagent G3 | 1.25 µL | 70.3 µL | 140.6 µL |
| Enzyme G4 | 0.50 µL | 28.13 µL | 56.26 µL |
| Enzyme G5 | 0.50 µL | 28.13 µL | 56.26 µL |
- Denature the 96-well plate in a thermocycler at 98 °C for 3 minutes. Immediately place on ice
to prevent rehybridization. Plate is ready once bottom of plate is cool to the touch. Quick spin plate 5s at 500xg before unsealing.
-
Transfer 10.5 μL Adaptase mix to sample plate. Seal, vortex, and quick spin (5s at 500xg).
-
Incubate plate in a thermocycler with the following program:
-
37°C for 30 minutes
-
95°C for 2 minutes
-
Hold at 4 °C
-
PCR Reaction:
-
Add 5 μL PCR primers to sample plate. Be sure each set of 6 wells (the original 384-well plate) gets a uniquely indexed primer.
-
Transfer 25 μL KAPA to each well. Seal, vortex, and quick spin (5s at 500xg).
-
Incubate plate in a thermocycler using the following method:
-
95°C for 2 minutes
-
98°C for 30 seconds
-
98°C for 15 seconds
-
64°C for 30 seconds
-
72°C for 2 minutes
-
Repeat steps c through e for a total of 13 cycles
-
72°C for 5 minutes
-
Hold at 4°C
-
**Note: 13 cycles is sufficient for mouse/human samples. More cycles should be run for smaller
genomes (eg. Arabidopsis)
4. Plate can be held at 4°C overnight or stored at -20°C for longer times
Final bead cleanups and pooling
Perform bead cleanup on PCR plate:
-
Thoroughly mix Sera-Mag beads by repeatedly inverting bottle and pour beads to the brim of the bead reservoir.
-
Add 40 μL beads to the sample plate and mix by pipetting. Upon addition of beads, start
a 5-minute timer.
-
When 5-minute timer is complete, move plate onto magnet. Wait until the beads are settled to continue (approximately 3 minutes).
-
Carefully remove supernatant and discard in the waste trough.
-
Perform two 180 μL EtOH washes on plate.
-
After second EtOH wash is complete, remove plate from the magnet.
-
Once beads are dry, add 20 μL EB to each well of the plate. Start a 5-minute timer.
-
Seal plate, vortex until beads no longer cling to sides of wells, and quick spin for 5s at 100xg.
-
After 5-minute timer is complete, place plate onto magnet and carefully unseal.
Perform bead cleanup on compressed samples in tubes:
- Using a 200 μL 8-channel pipette, compress wells such that samples from each
original 384-well plate are contained in a single well.
1. Sample from A1-A6 should end up in A6, sample from B1-B6 should end up in B6, etc.
2. Using a P-200 pipette, transfer sample from each well into an individually labeled, 1.5 mL microcentrifuge tube (16 tubes for 16 original 384-well plates).
-
Add 96 μL Sera-Mag beads to each microcentrifuge tube. Set a 5-minute timer, then vortex and quick spin all tubes.
-
After the 5-minute timer has completed, place each tube onto the DynaMag tube magnet. Allow beads to settle (approximately 3 minutes).
-
Remove and discard supernatant, careful to not disturb the beads.
-
Wash beads with 300 μL EtOH. Remove and discard EtOH.
-
Repeat step 6.
-
Using a P-10 pipette, remove any residual EtOH at the bottom of the tube and discard.
-
Remove tubes from DynaMag tube magnet and allow to dry with lid open until beads are visibly cracked and there are no visible droplets of EtOH.
-
Add 20 μL EB to each tube. Set a 5-minute timer, then vortex and quick spin all tubes.
-
After 5-minute timer has completed, place each tube onto the DynaMag tube magnet.
-
Transfer 20 μL from each sample tube into a new clean, labeled, 1.5 mL microcentrifuge tube. Try not to transfer any beads.
*TIP: It helps to not push the tubes all the way down on the magnet. Slide them in about halfway so the bead pellet stays near the liquid.
DNA Quantification and sample pooling:
-
To quantify the DNA in each sample tube, prepare an appropriate volume of Qubit reagent in a 15 mL microcentrifuge tube according to manufacturer’s instructions.
-
Label one Qubit assay tube per sample. Label two additional tubes for the standards
included in the Qubit kit. Also label tubes for any pools to be made. -
Combine Qubit reagent and DNA in Qubit assay tubes according to manufacturer’s
specifications.
-
-
Measure the concentrations using the Qubit.
-
Create sequencing pool by combining normalized amounts of each sample tube.
-
Check the concentration of the pool using the Qubit. Pool is now ready for sequencing.
