Use of the waxworm Galleria mellonella larvae as an infection model to study Acinetobacter baumannii
Kah Ern Ten, Nazmul Hasan Muzahid, Sadequr Rahman, tan.hocksiew
Galleria mellonella
Acinetobacter baumannii
infection
killing assay
bacterial burden assay
RNA extraction
Abstract
Galleria mellonella larvae have been increasingly used in various scientific research, including microbial infection studies. They act as suitable preliminary infection models to study host-pathogen interactions due to their advantages, such as the ability to survive at 37 °C mimicking human body temperature, their immune system shares similarities with mammalians, etc . Here, we presented a protocol for simple rearing and maintenance of Galleria mellonella in science research laboratories without requiring special instruments and specialised training. This allows the continuous supply of healthy Galleria mellonella for research purposes. Besides, this protocol also provides detailed procedures on the (i) Galleria mellonella infection assays (killing assay and bacterial burden assay) for virulence studies and (ii) bacterial cell harvesting from infected larvae and RNA extraction for bacterial gene expression studies during infection. Our protocol could not only be used in the studies of A. baumannii virulence but can also be modified according to different bacterial strains.
Attachments
Steps
Galleria mellonella rearing and maintenance
Research-grade Galleria mellonella larvae were ordered in bulk from Carolina Biological (US).
Set up Galleria mellonella housing according to Figure 2.
Fill 2/3 of the larvae jar with the freshly prepared medium.
Ingredients of artificial diet (per jar):
| A | B |
|---|---|
| NESTLE CERELAC® Infant Cereals Multi Grain & Garden Vegetables (Nestlé Malaysia) | 83.3 g |
| pure honey | 20 g |
| 99.8% glycerol | 20 g |
| instant baker yeast | 2.3 g |
Mix well in a clean plastic container using a spatula.
Transfer healthy larvae from the container provided by the vendor individually into a new glass jar with fresh medium, and cover with a layer of cloth-type voile.
Place the glass jars above a heating mat with temperature controlled at 32°C ± 2°C with humidity 44%-54% and keep them in a plastic storage box in a dark environment.
Add the fresh medium every 3 days and remove the sick/dead larvae from the jars to prevent the spread of diseases.
Dead/sick worms should be placed in a Petri dish or plastic bag and frozen at -20°C . Discard as biological waste.
Larvae should be transferred individually to a new fresh medium when the old medium is dirty.
Allow larvae to grow into the last instar stage (approximately 300 mg, 3 cm long). At this stage, no food is needed.
Transfer healthy, creamy-white larvae at the 6th instar stage to a new glass jar with fresh food and kept at Room temperature in a dark environment for experimental use.
Transfer 50 pupae (brownish colour) or larvae in the pre-pupal stage (with thick cocoons) into a moth jar using blunt-end forceps and cover with filter paper and a perforated lid.
Female moths will lay eggs around the filter paper.
Replace the filter paper (that has eggs on it) with a new filter paper. This should be performed every 3 days to avoid the escape of newly hatched larvae.
The moth jars should be cleaned after 2 weeks the first moth appeared to avoid the escape of newly hatched larvae. This can be done by placing the moth jars in a cold room (4°C) . Transfer the moths into a plastic bag and freeze them at -20°C . Discard as biological waste.
Cut the collected filter paper (with eggs) into smaller pieces and transfer it into the egg jar with food.
- Discard areas with contamination.
- Cover the egg jar with a perforated lid.
Note
NOTE :Extreme care is needed as the eggs can easily burst. Applying a layer of Vaseline® petroleum jelly at the wall of the egg jars is highly recommended to prevent the newly hatched larvae from escaping.
Egg jars should be monitored every 3 days to ensure a continuous food supply until they grow into the adult stage.
Separate the medium into half using blunt-end forceps and put it into two jars when it is too crowded. Top up the medium with freshly prepared food.
Transferring the larvae individually into a new jar with freshly prepared food might be necessary when the medium has fungal contamination or an unusual smell.
When the larvae grow bigger, transfer medium and large larvae (approx. 1.5 cm and 2 cm) to a new jar with food and cover with cloth-type voile and perforated lids.
Galleria mellonella infection assays: Sample preparation
Incubate 10 randomly chosen healthy 6th instar stage larvae (200-300 mg) with creamy-white appearance and no melanisation at 37°C, without food, in a standard bacterial incubator for one day before the experiment.
Pre-incubation at 37°C allows the selection of more suitable larvae, where unhealthy larvae will show melanisation and/or death after the pre-incubation and will be excluded from the experiment.
Prepare bacterial overnight culture by inoculating 1 colony of Acinetobacter baumannii in 5mL of Luria Bertani broth and incubate with shaking at 200rpm for 16-18 hours.
Cut pipette tips can be prepared by following Fredericks, Lee (2), which will be used as larvae restraint devices.
Sterilise the cut pipette tips by immersing them in 70% ethanol , then discarding the ethanol and autoclaving.
Galleria mellonella infection assays: Killing assay
Pellet 1mL of the overnight bacterial culture by centrifuging at 8300rpm.
Resuspend the bacterial pellet with 1mL of sterile 1X phosphate buffer saline (PBS) (7.4).
Repeat centrifugation (8300rpm) to pellet the bacterial culture.
Resuspend the bacterial pellet with 1mL of sterile 1X PBS (7.4).
Measure the optical density of the bacterial culture and adjust it to the appropriate OD600nm.
Wash the Hamilton syringe (model 725LT).
Wash the Hamilton syringe (model 725LT) with diluted bleach.
Then, wash it with distilled water to remove the bleach. (1/2)
Wash it with distilled water to remove the bleach. (2/2)
Attach the needle (27G) to the syringe and attach the Hamilton syringe to the Hamilton repeating dispenser (PB600-1).
Sterilise the larval prolegs with 70% ethanol using a cotton swab.
Place the larval tail into the wider part of the cut tip, then insert the narrow part of the cut tip to trap the larvae.
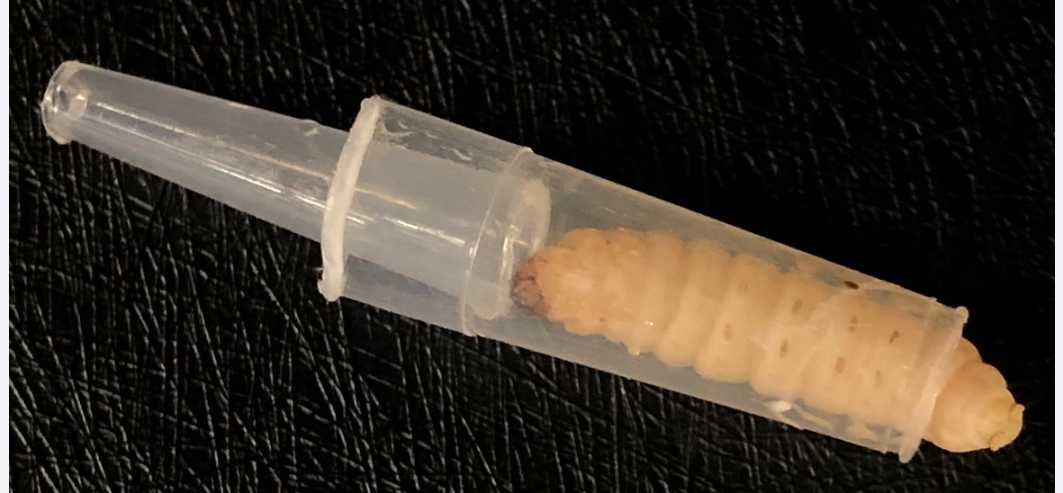
Inject 10µL of bacterial suspensions with desired cell density into the last left proleg of larvae.
The needle should be visible through the larval cuticle after inserting it into the proleg.
The Hamilton syringe should be cleaned after each experimental group (step 20) to avoid being carried over to the next experimental group.
Two control groups should be used:
- larvae injected with only sterile PBS (to assess physical trauma),
- larvae without receiving any injections (non-manipulated control to assess background mortality).
Place the larvae in a sterile Petri dish lined with filter paper.
Incubate the larvae at 37°C in a standard bacterial incubator and score for survival every 24h 0m 0s.
Larvae are considered dead when they are unresponsive to physical stimuli and melanised.
Remove larvae from the cocoon to check survival, and dead larvae should be removed from the plate at every time point to avoid the spread of diseases.
Stop the experiment when pupation occurs to avoid biases.
Repeat the experiments independently 3 times to get the data of 3 biological replicates ( n =30).
Perform the Kaplan-Meier survival curves and statistical analysis (log-rank test) using GraphPad Prism software.
Galleria mellonella infection assays: Bacterial burden assay (quantification of bacterial CFU in vivo)
Adjust overnight bacterial culture (washed twice with sterile 1X PBS, 7.4) to appropriate OD600 nm. Inoculum is always confirmed via plating.
Sterilise the larval prolegs with 70% ethanol using a cotton swab.
Trap the larvae in the restraint devices.
Clean the Hamilton syringe with diluted bleach and distilled water.
Inject 10µL of bacterial suspension into the last left proleg of the larvae. Place the larvae in a sterile Petri dish lined with filter paper and incubate at 37°C in the standard bacterial incubator.
Negative control group: larvae injected with sterile 1X PBS only.
Measure and record the weight of a sterile microcentrifuge tube (1.5 mL) before the hemolymph collection.
At each time point, randomly choose 3 larvae from the incubated larvae.
Anaesthetise them On ice in a 15 mL centrifuge tube for 0h 10m 0s.
Sterilise the larval surface by immersing them in 70% ethanol for 0h 0m 30s, followed by washing.
Washing with sterile distilled water to remove the residual ethanol. (1/2)
Washing with sterile distilled water to remove the residual ethanol. (2/2)
Make an incision by puncturing the cuticle between the second and third proleg using a sterile 27G Terumo needle. Squeeze the larvae with sterile plastic forceps (sterilised with diluted bleach and 70% ethanol) and collect the hemolymph immediately from the puncture site via pipetting.
Pool the hemolymph from 3 larvae into the weighted microcentrifuge tube.
Incubate with 1µL of digitonin (5mg/mL) at Room temperature to lyse haemocytes and release intracellular bacteria (3).
Perform 10-fold serial dilution on the collected hemolymph and plate 100µL of the bacteria with appropriate dilution onto Leeds Acinetobacter agar.
Incubate the agar plate at 37°C for 20h 0m 0s.
Count the number of bacterial colonies and calculate the CFU/larva by normalising to the weight of hemolymph extracted.
Perform the experiments in three independent replicates.
Plot the bacterial growth curve in vivo using GraphPad Prism software.
Bacterial harvesting and RNA extraction from infected larvae: Sample preparation
Inoculate a single colony of bacteria in 5mL of Luria-Bertani broth and incubate for 16-18 hours with continuous shaking at 200rpm.
Incubate 40 healthy larvae (6th instar stage, 200-300 mg) with a creamy-white appearance at 37°C 20h 0m 0s.
Bacterial harvesting and RNA extraction from infected larvae: Bacterial cell harvesting from infected larvae
Spin 1mL of the overnight bacterial culture at 8300rpm.
Resuspend in 1mL sterile 1X PBS.
Spin the bacterial culture again at 8300rpm, and resuspend in 1mL sterile 1X PBS.
Adjust the bacterial culture to the appropriate optical density. Inoculum is always confirmed via plating.
Sterilise the larval prolegs with 70% ethanol using a cotton swab.
Inject 10µL of bacterial suspension into the last left proleg of the larvae using a Hamilton syringe (model 725LT) with a 27G Terumo needle.
Place the larvae in a sterile Petri dish lined with filter paper and incubate at 37°C in the standard bacterial incubator for 3h 0m 0s.
During the incubation time, prepare a stop mix solution (95% absolute ethanol: 5% Tri-RNA) in a 2 mL RNase-free microcentrifuge tube and keep it at -20°C. Pre-cool microcentrifuge to 4°C.
At the desired time point, sterilise the larval surface by immersing the larvae in 70% ethanol for 0h 0m 30s, followed by rinsing two times with sterile distilled water.
Extract the hemolymph from the infected larvae using a Terumo 27G needle by puncturing the larval cuticle between the second and the third prolegs. Collect the hemolymph immediately from the punctured site and pool it into the microcentrifuge tube with ice-cold 0.2 volume of stop mix solution.
Incubate the hemolymph-stop mix solution mixture at Room temperature for 0h 5m 0s.
Gently vortex for 0h 0m 5s.
Centrifuge at 2300rpm,4°C. Collect the supernatant and transfer it into a 1.5 mL RNase-free microcentrifuge tube.
Repeat step 58 until no or very little host pellet is obtained.
Pellet the bacterial cells from the supernatant by centrifugation at 10000rpm,4°C. Discard the supernatant.
Immediately resuspend the bacterial cell pellet in 1mL of tri-RNA. Homogenise the sample by gentle vortex for 0h 0m 5s or via repetitive pipetting.
Store at -80°C or proceed to RNA extraction.
Bacterial harvesting and RNA extraction from infected larvae: Bacterial RNA extraction
Thaw the sample On ice.
Pre-cool the microcentrifuge to 4°C.
Incubate at Room temperature for 0h 5m 0s.
Add 200µL of chloroform into the mixture. Mix by inversion until a milky pink mixture is obtained.
Incubate at Room temperature for 0h 5m 0s.
Centrifuge at 13000rpm,4°C.
Collect the aqueous transparent supernatant into a 2 mL RNase-free microcentrifuge tube.
Add an equal volume of 95% ethanol (molecular grade).
Extract RNA according to the manufacturer’s manual (Monarch® Total RNA Miniprep Kit, NEB) as described below:
Load the mixture onto the RNA purification column.
Centrifugation at 16000x g. Discard the flowthrough.
On-column DNase I treatment:
- Add
500µLRNA Wash Buffer and centrifuge for0h 0m 30s. Discard flow-through. - Add DNase I mixture (
5µLDNase I with75µLDNase I Reaction Buffer) onto the column. - Incubate for
0h 15m 0satRoom temperature.
Add 500µL RNA Priming Buffer and centrifugate at 16000x g. Discard flow-through.
Add 500µL RNA Wash Buffer and centrifugate at 16000x g. Discard flow-through.
Add 500µL RNA Wash Buffer and centrifugate at 16000x g.
Transfer the column to a 1.5 mL RNase-free microcentrifuge tube.
Load 20µL of RNase-free water onto the column and incubate for 0h 5m 0s at Room temperature.
Centrifugation at 16000x g to elute the RNA.
Assess the RNA quality by gel electrophoresis (1% agarose gel) and Agilent TapeStation 2200 and measure the absorbance values (A260/230 and A260/280) and concentration using a BioDrop spectrophotometer.
Store the RNA at -80°C or place it On ice for immediate downstream applications.
