Top Down Proteomics Data Collection for Microdissected Pancreas Tissue Functional Units
Mowei Zhou, Ljiljana.PasaTolic, James M Fulcher, Isaac Kwame Attah
Abstract
The protocol describes how to use laser capture microdissection (LCM) to cut small regions of interest (~200-300 μm) from tissue sections. This is followed by top down proteomics analysis by liquid chromatography - mass spectrometry (LC-MS).
Steps
Liquid chromatography (LC) method setup
Set up reversed-phase LC system with online trapping for desalting.
Dual pump configuration
Mobile phase A (MPA): 0.2% formic acid in water (LCMS grade)
Mobile phase B (MPB): 0.2% formic acid in acetonitrile (LCMS grade)
Equipment
| Value | Label |
|---|---|
| NanoAcquity | NAME |
| liquid chromatography | TYPE |
| Waters | BRAND |
| 186016002 | SKU |
| http://www.waters.com/waters/home.htm?locale=en_DK | LINK |
| Dual pump configuration with autosampler 186016007 | SPECIFICATIONS |
Prepare the method for autosampler for microPOTS samples.
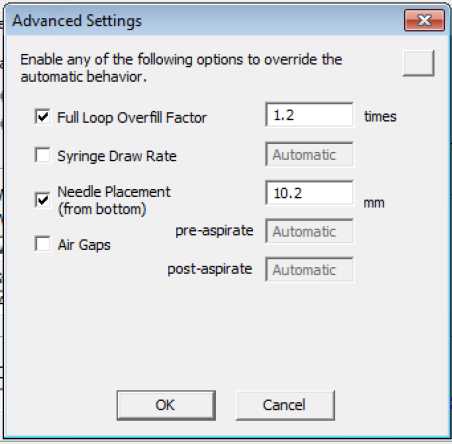
Set up gradient method for samples.
Wash pump: 5 μL/min, 95% MPA, 5% MPB. Loading time 10 min.
Gradient pump: 0.3 μL/min
0 min: 95% MPA, 5% MPB
1 min: 90% MPA, 10% MPB
90 min: 40% MPA, 60% MPB
100 min: 95% MPA, 5% MPB
Mass spectrometer (MS) method setup
Calibrate and set up the mass spectrometer method for sample runs.
Perform both mass and system calibration following instrument vendor's recommendation.
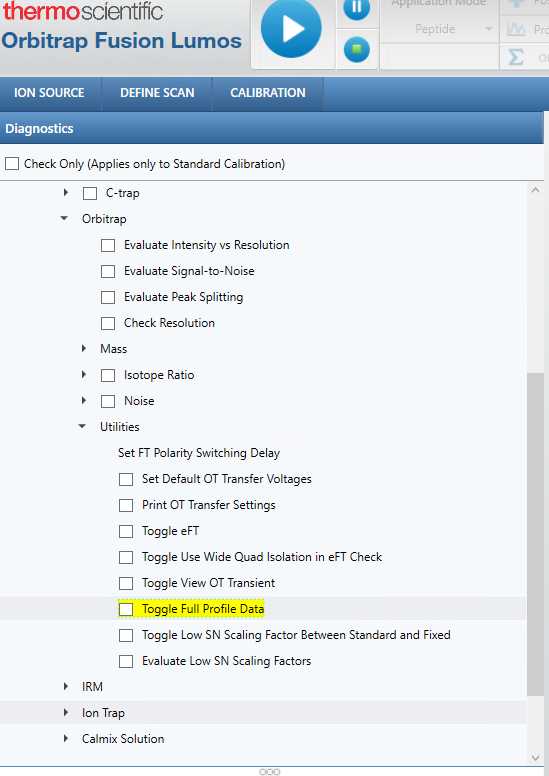
(Optional) Turn on full profile mode.
Under "Diagnosis" - "System" - "Orbitrap" - "Utilities", check "Toggle Full Profile Data" before starting the queue.
Set up the data dependent acquisition method with the following parameters.
ETD method was used to obtain higher sequence coverage than CID for characterizing proteoforms. In addition, a ETD method with inclusion list for histones was used on some replicates to improve the analysis of histone modifications.
Sample method - CID
Sample method - ETD
Histone target inclusion list (H4 and H3) for ETD method
Instrument Quality Control (QC) and method setup
A QC standard is used to evaluate instrument performance before starting samples.
Herein we use a bacterial lysate established in our lab (see reference below for more information), other samples can be used as QC as well.
QC Sample information
Intact protein lysate from cultured Shewanella oneidensis MR-1 cells
Buffer preparation :
Homogenization buffer ( HB ) : 8M urea solution (480 mg/mL) in 50 mM ABC with 15 mM TCEP
Note: Use BondBreaker 0.5 M TCEP stock solution
Wash Buffer ( WB ): 0.2% formic acid, 5% acetonitrile
Note: Use LC-MS grade water
- Lyse cells or homogenize tissue in homogenization buffer (HB).
- Incubate sample at room temperature for 30 min to extract and denature proteins
- Centrifuge lysate at 14,000 x G, 10C for 10 minutes to pellet cell debris
- Transfer supernatant to 100K MWCO filter and centrifuge at 14,000 x G until minimum volume is reached.
- Wash 100K spin filter with 1X max volume of HB, spin at 14,000 x G until minimum volume is reached.
- Transfer filtrate from 100K filter to a fresh 10K filter and centrifuge at 14,000 x G for time needed to get to minimum volume. a. If needed, add multiple aliquots of filtrate from 100K filter to the same 10K filter
- Wash 10K filter three times with wash buffer (WB) and spin to minimum volume each wash.
- Perform Coomassie or BCA protein assay.
- Dilute sample to 0.01 ug/uL in WB and aliquot 100 uL into separate 0.6 mL Eppendorf tubes with labels.
QC LCMS method
LC method
Wash pump: 5 μL/min, 95% MPA, 5% MPB. Loading time 15 min.
Gradient pump: 0.3 μL/min
0 min: 95% MPA, 5% MPB
180 min: 55% MPA, 45% MPB
Followed by a blank injection to wash the column before sample runs
MS method
Data collection and QC metric
Queue the QC sample before starting the samples using the LCMS method described in section 3. QC runs need to pass the metrics defined below. Once passed, queue the sample runs using the LCMS method described in section 2.
Evaluation of QC data ( Shewanella lysate from section 3)
To quickly evaluate the QC data, open them in the Thermo Scientific Freestyle software or Xcalibur QualBrowser. The following metrics are inspected to ensure that the LCMS run meets the expected standard.
1. Signal levels
The intensity levels at the total ion current (TIC), base peak intensity (BPI), and the MS2 spectra are inspected to ensure that they meet the expected intensity levels. The TIC intensity levels are typically expected to be above the 1e10 level, BPI at or above 1e8 level, and the MS2 spectra ion current from 1e5 to 1e7 levels.
2. Chromatography
2a.
We look at the chromatography distribution using the base peak FTMS scan as filter to ensure that the eluted peaks are well distributed through the LC run. The chromatogram is filtered to show the BPI peaks and inspected to ensure no peak broadening (early or late) is observed.

2b.
The distribution of the MS2 peaks (fragmentation of peaks selected at the MS1 level) is inspected to evaluate how similar the distribution is to that of the MS1 level. The MS2 distribution is expected to emulate what is observed for the peak distribution of the MS1 when the BPI filter is applied, indicating that peak selection for fragmentation was performed at an appreciable level. To assess the MS2 fragmentation signal, the “full MS2” is applied as the filter to show the MS2 distribution through the whole experiment.
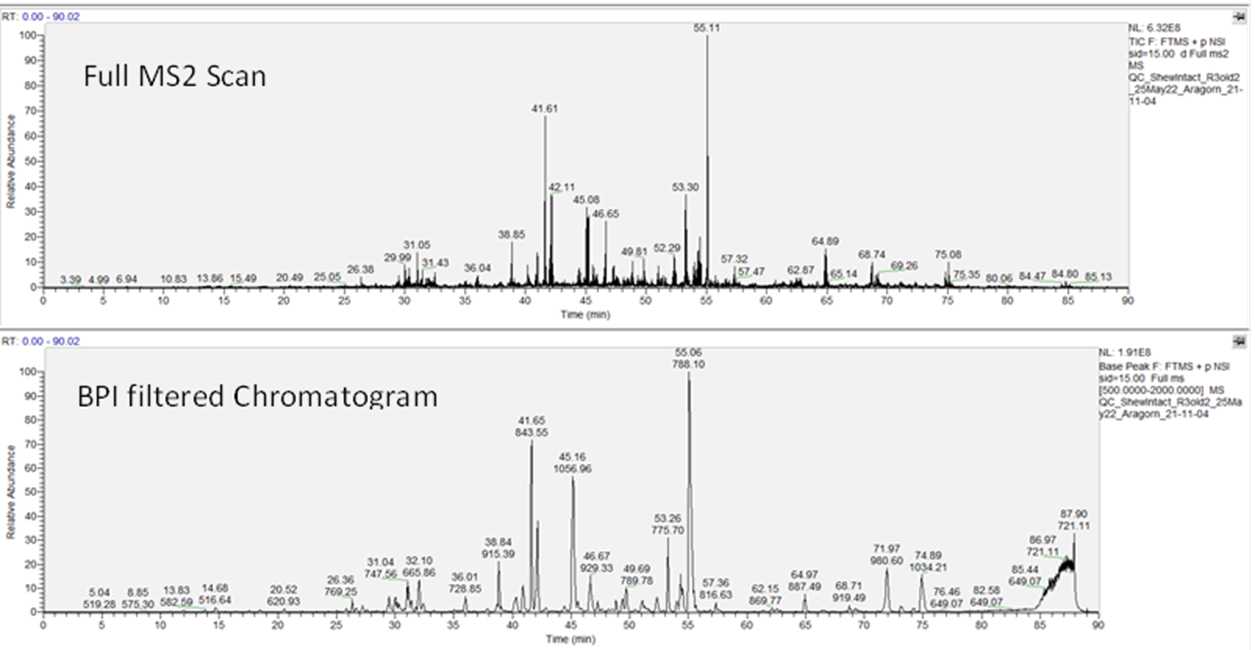
2c.
Finally, QC samples are analyzed with TopPIC to ensure appropriate number of proteoforms and proteoform spectrum matches (PrSMs) are being identified.
Software
| Value | Label |
|---|---|
| TopPIC Suite | NAME |
| https://github.com/toppic-suite/toppic-suite | REPOSITORY |
| Xiaowen Liu | DEVELOPER |
| https://www.toppic.org/software/toppic/index.html | LINK |
| 1.4.13.1 | VERSION |
Proteoforms are counted by opening the exported "..._proteoforms.tsv" file and PrSMs through the "..._PrSMs.tsv" file. The QC passing threshold for proteoforms is 1,000 and 2,000 for PrSMs.
Quality Assurance (QA) of HubMAP Samples
Perform proteoform identification using the "TopPIC processing" section in the following protocol.
Using the TopPIC PrSM results (after TopPICR post-processing), kidney samples are filtered based on the total number of PrSMs. A cutoff of 100 PrSMs was used to remove samples that were of lower quality.
