QIAGEN QIAseq FX Protocol
Emily Gailey
Abstract
QIAseq Normalizer Kits provide a fast and easy way to normalize and pool NGS libraries for
Illumina, while skipping library qualification and quantification steps. QIAseq Normalizer will
accurately normalize over a broad range of library concentrations, but it cannot fully
compensate for libraries of poor quality or libraries at lower concentrations of less than
15 nmol/L.
Steps
Procedure
Program a thermocycler according to Table 2 using the predetermined FX fragmentation
time for step 2. Be sure to use the instrument’s heated lid, and if possible, set the
temperature of the heated lid to 70°C.
Table 2. Input DNA (20 pg –1000 ng) free of EDTA, Buffer EB, or in 0.1x T

Start the program. When the thermocycler block reaches 4°C, pause the program.
Prepare the FX reaction mix in a PCR plate or tube on ice according to Table 3 for >10 ng
input DNA or Table 4 for <10 ng input DNA. Mix well by gently pipetting (do not vortex
to mix).
Table 3. FX reaction mix setup (per sample) for >10 ng input DNA

Table 4. FX reaction mix setup (per sample) for <10 ng input DNA

Add 10 µL FX Enzyme Mix to each reaction and mix well by pipetting up and down
20 times. It is critical to keep the reactions on ice for the entire time during reaction setup.
Briefly spin down the PCR plate/tubes and immediately transfer to the prechilled
thermocycler (4°C). Resume the cycling program.
When the thermocycler program is complete and the sample block has returned to 4°C,
remove samples and place them on ice.
Immediately proceed with adapter ligation.
Pierce the foil seal for each adapter well to be used, and transfer 5 µL from one DNA
adapter well to each 50 µL sample from the previous protocol. Track the barcodes from
each adapter well used for each sample.
Note : If your DNA input is <10 ng, dilute the adapters according to Table 5.
Table 5. Adapter dilution factors

Freeze the adapter plate containing unused adapters. QIAseq adapters are stable for a
minimum of 10 freeze-thaw cycles.
Important : Only 1 single adapter should be used per ligation reaction. If adapters from
another supplier are used, follow the manufacturer’s instructions. Do not reuse adapter
wells once the foil seal has been pierced.
Prepare the ligation Master Mix (per DNA sample, Table 6) in a separate PCR plate or
tube on ice, and mix well by pipetting.
Table 6. Ligation master mix setup (per sample)

Add 45 µL of the ligation Master Mix to each sample, for a total of 100 µL, and mix well
by pipetting. Incubate the ligation reaction at 20°C for 15 min. Important nt: Do not use a thermocycler with a heated lid.
Proceed immediately to adapter ligation cleanup using 0.8x (80 µL) Agencourt AMPure
XP beads or QIAseq Beads.
Add 80 µL of resuspended Agencourt AMPure XP beads or QIAseq Beads to each ligated
sample and mix well by pipetting.
Incubate the mixture for 5 min at room temperature. Pellet the beads on a magnetic stand
(e.g., DynaMag) for 2 min, then carefully discard the supernatant.
Wash the beads by adding 200 µL of 80% ethanol. Pellet the beads on the magnetic stand
and discard the supernatant. Repeat the wash once, for a total of 2 ethanol washes.
Remove as much excess ethanol as possible.
Incubate the beads on the magnetic stand for 5–10 min or until the beads are dry.
16a. Overdrying of Ampure XP beads may result in lower DNA recovery.
16b. If using QIAseq Beads, ensure that the pellet is completely dry by visual inspection.
Over drying QIAseq Beads will not affect the DNA elution.
Remove from the magnetic stand. Elute by resuspending in 52.5 µL of Buffer EB or 10 mM
Tris·Cl, pH 8.0. Pellet the beads on the magnetic stand. Carefully transfer 50 µL of
supernatant into a new plate or tube.
Perform a second purification using 1x (50 µL) Agencourt AMPure XP beads or 1.1x (55 µL)
of QIAseq Beads following steps 14–16 for DNA binding and washing. Elute DNA by
adding 26 µL Buffer EB or 10 mM Tris·Cl, pH 8.0. Pellet the beads and carefully collect
23.5 µL of purified DNA sample in a DNA LoBind tube for library amplification. If not
proceeding immediately, the sample can be stored at −30°C to −15°C.
Amplification of Library DNA
Program a thermocycler with a heated lid according to Table 7.
Table 7. Library amplification cycling conditions

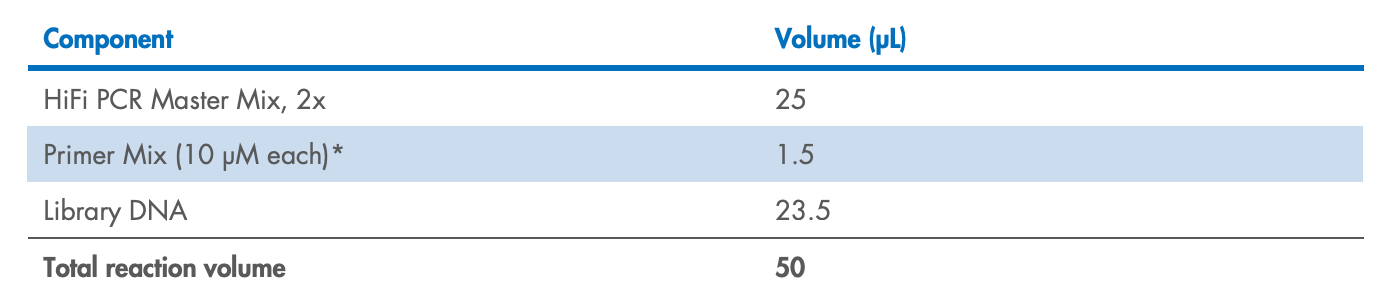
Prepare a reaction mix on ice according to Table 8. Mix the components in a PCR tube or
96-well PCR plate.
Table 8. Reaction mix for library enrichment
Transfer the PCR tube or plate to the thermocycler and start the program.
Once PCR is complete, add 50 µL of resuspended Agencourt AMPure XP Beads or 60 µL
of resuspended QIAseq Beads to each reaction (50 µL) and pipette up and down
thoroughly to mix.
Incubate the mixture for 5 min at room temperature. Pellet the beads on a magnetic stand
(e.g., DynaMag) and carefully discard the supernatant.
Wash the beads by adding 200 µL of 80% ethanol. Pellet the beads on the magnetic stand
and discard the supernatant. Repeat the wash once, for a total of 2 ethanol washes.
Remove as much excess ethanol as possible.
Incubate on the magnetic stand for 5–10 min or until the beads are dry.
25a. Overdrying of Ampure XP beads may result in lower DNA recovery.
25b. If using QIAseq Beads, ensure that the pellet is completely dry by visual inspection.
Overdrying QIAseq Beads will not affect the DNA elution.
Remove from the magnetic stand. Elute by resuspending in 25 µL of Buffer EB or 10 mM
Tris·Cl, pH 8.0. Pellet the beads on the magnetic stand. Carefully transfer 23 µL of the
supernatant into a new tube.
26a. Overdrying of Ampure XP beads may result in lower DNA recovery. Remove from
the magnetic stand.
26b. If using QIAseq Beads, ensure that the pellet is completely dry by visual inspection.
Overdrying QIAseq Beads will not affect the DNA elution.
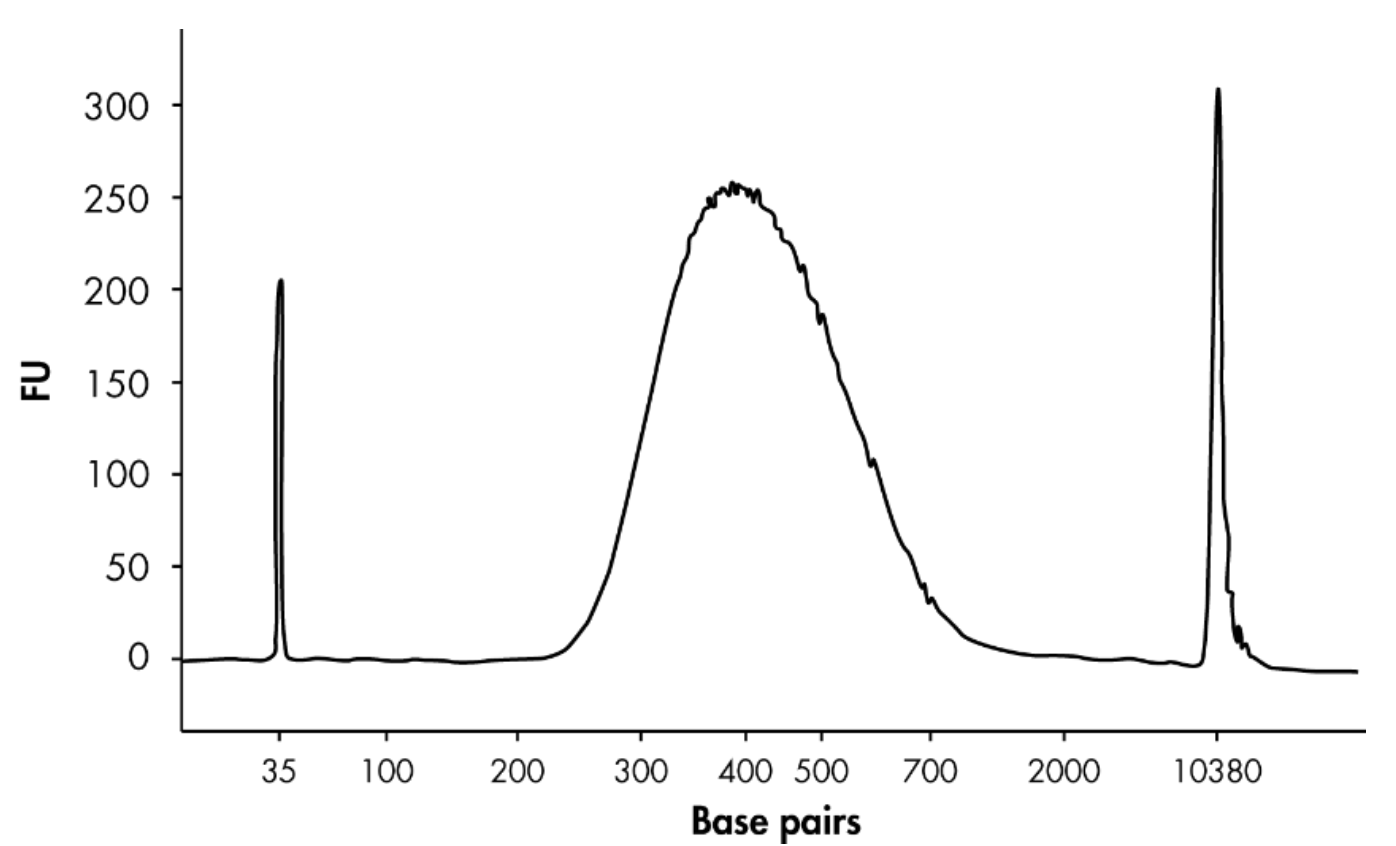
Assess the quality of the library using a capillary electrophoresis device such as QIAGEN
QIAxcel or Agilent BioAnalyzer. Check for the expected size distribution (see Figure 2) of
library fragments and for the absence of an adapter-dimer peak around 120 bp Note ote: The library should show a distribution centered around the size of the fragmented
DNA plus 120 bp. The increase in library length reflects the
addition of sequencing adapters to the DNA fragme Note
Note: The median fragment size can be used in subsequent qPCR-based quantification
methods to calculate library concentration (step 28).
Quantify the library using a qPCR-based method such as the QIAseq Library Quant Assay
Kit (cat. no. 333314; not provided), or a comparable method.
The purified library can be safely stored at −30°C to −15°C in a DNA LoBind tube until
ready to use for sequencing or other applications.
Figure 2. Capillary electrophoresis device trace data.