Metabarcoding using MinION: PCR, Multiplexing and Library Preparation
Bastian Egeter, Joana Veríssimo, Manuel Lopes-Lima, Cátia Chaves, Joana Pinto, Nicoletta Riccardi, Pedro Beja, Nuno A. Fonseca
Abstract
A protocol for the metabarcoding of DNA samples using nanopore technology, for the purposes of biomonitoring, biodiversity assessment, DNA-based diet analysis, or other related applications.
The protocol is being used in our laboratory for detecting aquatic invasive bivalve species and for measuring biodiversity of fish and other vertebrates from water eDNA samples, as well as for species identification of scats.
The key steps are a first PCR to amplify metabarcoding fragments, a 2nd PCR to add indexes (barcodes) to allow pooling of multiple samples, and the preparation of the pool for sequencing on a MinION sequencer.
The protocol is adapted from PCR barcoding (96) amplicons (SQK-LSK109) by Nanopore.
Before start
Note: Steps for DNA extraction and associated DNA quality checks are outside the scope of this protocol. It is assumed that suitable DNA samples are being used as input for the initial PCR.
Also, note that this protocol is optimised for fragments <500bp (using Qiagen Master Mix for the barcoding PCR reaction). If targeting longer amplicons (particularly over 1kb), modification of steps 3-5 should be considered, e.g. using LongAmp Taq 2x Master Mix (New England Biolabs [NEB] USA).
Steps
Preparing first PCR
Prepare first PCR with a 25µL total PCR volume using optimised conditions for the primers / enzyme in use. Primers should include MinION adapters (5´-TTTCTGTTGGTGCTGATATTGC-forward primer-3´, 5´-ACTTGCCTGTCGCTCTATCTTC-reverse primer-3´).
Test PCR product to assess amplification success, e.g. via gel electrophoresis with 2 % agarose gels stained with GelRed (Biotium, USA).
Barcoding PCR: 96-BARCODING KIT (EXP-PBC096)
In a 0.2 mL 96-well plate, set up a barcoding PCR reaction for each library, as follows:
1µL
24µL
25µL
Mix by pipetting.
Amplify, in a thermal cycler (e.g. T100 Thermal Cycler, BioRad, USA), using the following cycling conditions:
- Initial denaturation
0h 15m 0s@95°C(1 cycle), - Denaturation
0h 0m 15s@95°C(12 cycles), - Annealing
0h 0m 15s@62°C(12 cycles), - Extension
0h 0m 30s@72°C(12 cycles), - Final extension
0h 10m 0s@72°C(1 cycle), - Hold @
4°C.Safety informationSuitable stopping point. Can store sample at -20° C long-term.
Pool 5µL of each sample into a single 1.5 mL tube. If multiple primers were used in the first PCRs, and libraries from the different primer sets will be combined in the same sequencing run, we suggest preparing a separate pool for each primer set.
- Note that this may result in substantial variation in the number of reads/sample obtained, particularly if amplicon concentrations are highly variable. If aiming to reduce such variation, it is recommended to purify each PCR product using bead purification, calculate the concentration of each PCR product (e.g. using spectrophotometry or fluorometry), and normalise all samples to be equimolar prior to pooling. Note that amplicon length increases c. 94bp during the previous step. 48bp are the barcodes themselves (24 X 2bp), we assume the other 46bp are flanking adapter sequences.
Use automated electrophoresis (e.g. 2200 Tapestation System using a D1000 assay; Agilent Technologies, USA) to perform fragment analysis of each pooled library.
Bead Purification Protocol for each pooled library (adjust ratio of beads according to fragment lengths observed in previous steps, use a ratio of 0.6 X to clean fragments <300 bp):
- Prepare the Agencourt AMPure XP beads (Beckman Coulter, USA) for use (30 minutes at RT - room temperature) and freshly prepared 70% ethanol.
- Resuspend AMPure XP beads by vortexing.
- Transfer
100µLof the pooled library to a clean 2 mL Eppendorf tube (adjust this volume if low number of samples used). - Add
60µLof resuspended AMPure XP beads and mix by flicking the tube or pipetting up and down 10 times. - Incubate for 7-10 minutes at RT and invert occasionally for mixing.
- Spin down the tube and place it on a magnetic rack (e.g. DynaMag-2 Magnet; Thermo Fisher Scientific, USA) for 5 mins (or until supernatant is clear).
- Keeping the tube on the magnet, pipette off the supernatant and discard.
- Taking care not to disturb the beads, add
200µLof 70% ethanol, then pipette off the ethanol and discard. - Repeat the previous step.
- Spin down the tube and place the tube back on the magnetic rack. Pipette off any residual ethanol. Allow to air dry at RT for ~30 seconds.
- Remove the tube from the magnetic rack and resuspend the pellet in 80 µL nuclease-free water. Incubate for 2 minutes at RT.
- Place the tube back on the magnetic rack for 5 min (or until the eluate is clear and colourless).
- Transfer
70µLof eluate into a clean 1.5 mL Eppendorf tube.
Quantify each cleaned pooled library using fluorometer assay (e.g. Qubit dsDNA HS or BR assay kit; Thermo Fisher Scientific, USA).
If multiple pooled libraries were made, combine the pools in the ratios desired for sequencing, e.g. if twice as many reads are desired for Pool1 compared to Pool2, combine the pools so that Pool1 has twice the DNA mass of Pool2.
Prepare 1 μg of final pooled library in 47µL Nuclease-free water:
- The volume of the final pooled library to take equals 1000 divided by the pooled library concentration (ng/µL)
- Use nuclease-free water to bring final volume to
47µLSafety informationSuitable stopping point. Can store sample at -20° C long-term.
DNA repair and end-prep (SQK-LSK109)
Prepare the AMPure XP beads for use (30 minutes at RT) and prepare fresh 70% ethanol.
Thaw DNA CS (DCS, Ligation Sequencing Kit SQK-LSK109,ONT, UK) at RT, spin down, mix by pipetting, and place on ice.
Place NEBNext FFPE Repair Mix (NEB, USA) and NEBNext Ultra II End Repair/dA-Tailing Module (NEB, USA) on ice.
In a 0.2 mL thin-walled PCR tube, mix the following:
1µL
47µL
3.5µL
2µL
3.5µL
3µL
Mix gently by flicking the tube, and spin down.
Using a thermal cycler, incubate at 20°C for 0h 7m 0s and 65°C for 0h 7m 0s, with lid heating turned off.
Repeat Bead Purification Protocol (see ), starting with the full 60µL from the previous step and using 60µL beads (here this is a 1 X ratio). Use 61µL for final resuspension.
Adapter ligation and clean-up
Spin down Adapter Mix (AMX, Ligation Sequencing Kit SQK-LSK109, ONT, UK) and T4 Ligase (Quick Ligation Module NEB, USA), and place on ice.
Thaw Ligation Buffer (LNB, Ligation Sequencing Kit, SQK-LSK109,ONT, UK) at RT, spin down and mix by pipetting. Due to viscosity, vortexing this buffer is ineffective. Place on ice immediately after thawing and mixing.
Thaw Elution Buffer (EB, Ligation Sequencing Kit, ONT, UK) at RT, mix by vortexing, spin down and place on ice.
To retain DNA fragments shorter than 3 kb (by purifying fragments of all sizes), thaw one tube of S Fragment Buffer (SFB, Ligation Sequencing Kit, SQK-LSK109, ONT, UK) at RT, mix by vortexing, spin down and place on ice. If targeting longer fragments, use Long Fragment Buffer (LFB).
In a 2 mL Eppendorf tube, mix in the following order:
60µL
25µL
10µL
5µL
Mix gently by flicking the tube, and spin down.
Incubate tube for 10 minutes at RT.
Bead Purification with a ratio of 0.4 X ratio after adapter ligation:
- Prepare the AMPure XP beads for use; resuspend by vortexing.
- Add
40µLof resuspended AMPure XP beads, to tubes from previous step, and mix by flicking the tube. - Incubate for 7-10 minutes at RT and invert occasionally for mixing.
- Spin down the tube, place it on a magnetic rack for 5 mins (or until supernatant is clear).
- Keeping the tube on the magnet, pipette off the supernatant and discard.
- Add
250µLS Fragment Buffer (SFB) and flick the tube to resuspend, spin down the tube, place it on a magnetic rack for 5 mins (or until supernatant is clear). Keeping the tube on the magnet, pipette off the supernatant and discard. - Repeat previous step.
- Spin down the tube and place the tube back on the magnetic rack. Pipette off any residual S Buffer. Allow to air dry at RT for ~30 seconds.
- Remove the tube from the magnetic rack and resuspend the pellet in
15µLElution Buffer (EB). Incubate for 10 minutes at RT. For high molecular weight DNA, incubate at 37° C to improve the recovery of long fragments. - Spin down the tube, place it on a magnetic rack for 5 mins (or until supernatant is clear).
- Transfer
15µLof eluate to a clean 1.5 mL Eppendorf tube.
Quantify 1µL of eluate using a fluorometer assay (e.g. Qubit e.g. Qubit dsDNA HS or BR assay kit; Thermo Fisher Scientific, USA).
Using the concentration assessed in the previous step, calculate the molarity of the library (in fmol), using the formula below:
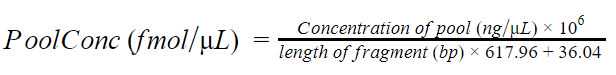
Dilute the library to 35 fmol (the range can vary from 5 - 50 fmol) in a final volume of12µL, using the concentration assessed previously. Use the formula below to assess how many μL from the pool you need.

Priming and loading the SpotON flow cell
Thaw the Sequencing Buffer (SQB, Ligation Sequencing Kit, ONT UK), Loading Beads (LB, Ligation Sequencing Kit, ONT UK), Flush Tether (FLT, Flow Cell Priming Kit, EXP-FLP001, ONT, UK) and one tube of Flush Buffer (FLB, Flow Cell Priming Kit, EXP-FLP001, ONT, UK) to RT before placing the tubes on ice as soon as thawing is complete.
Mix the Sequencing Buffer (SQB) and Flush Buffer (FLB) tubes by vortexing, spin down and return to ice.
Spin down the Flush Tether (FLT) tube, mix by pipetting, and return to ice.
Place the R9.4 flow cell (FLO-MIN106D, ONT, UK) in the MinION sequencer (Mk1B, ONT, UK).
Open the lid of the sequencer and slide the flow cell's priming port cover clockwise so that the priming port is visible.
After opening the priming port, check for small bubbles under the cover. Draw back a small volume to remove any air bubbles:
- Set a P1000 pipette to 200 µL,
- Insert the tip into the priming port,
- Turn the wheel until the dial shows 220-230 µL, or until you can see a small volume of buffer entering the pipette tip.
Prepare the flow cell priming mix: add 30µL of thawed and mixed Flush Tether (FLT) directly to the tube of thawed and mixed Flush Buffer (FLB), and mix by pipetting.
Load 800µL of the priming mix into the flow cell via the priming port, avoiding the introduction of air bubbles. Wait for 5 minutes.
Thoroughly mix the contents of the Loading Beads (LB) by pipetting.
In a new 1.5 mL Eppendorf tube, prepare the library for loading as follows:
37.5µL
25.5µL
12µL
Complete the flow cell priming:
- Gently lift the SpotON sample port cover to make the SpotON sample port accessible,
- Load
200µLof the priming mix into the flow cell via the priming port (not the SpotON sample port), avoiding the introduction of air bubbles.
Mix the sequencing pool gently by pipetting up and down just prior to loading.
Add 75µL of the sequencing pool to the flow cell via the SpotON sample port in a dropwise fashion. Ensure each drop flows into the port before adding the next.
Gently replace the SpotON sample port cover, making sure the bung enters the SpotON port, close the priming port and close the MinION lid.
The MinION is now ready to commence sequencing according to user specifications.
