MEDI: Macronutrient Extraction and Determination from Invertebrates
Jordan P Cuff, Shawn M. Wilder
macronutrient
nutritional
nutrient
invertebrate
insect
colorimetric assay
entomology
entomological
exoskeleton
lipid
protein
carbohydrate
Abstract
Macronutrients, comprising carbohydrates, proteins and lipids, underpin many ecological processes, but their quantification in ecological studies is often inaccurate and laborious, requiring large investments of time and bulk samples, which make individual‐level studies impossible. This is a protocol for the direct, rapid and relatively low‐cost determination of macronutrient content from single small macroinvertebrates.
Macronutrients are extracted by a sequential process of soaking in 1:12 chloroform:methanol solution to remove lipid and then solubilising tissue in 0.1 M NaOH. Proteins, carbohydrates and lipids were determined by colorimetric assays from the same individual specimens.
Macronutrient Extraction and Determination from Invertebrates (MEDI) can directly and rapidly determine macronutrient content in tiny (dry mass ~3 mg) and much larger individual invertebrates. Using MEDI, the total macronutrient content of over 50 macroinvertebrates can be determined within around 3 days of collection at a cost of ~$1.35 per sample.
Before start
Please read the full protocol before starting in order to decide which section are relevant to your study and how the overlapping aspects (if you decide to overlap them) should be followed.
Steps
Welcome to MEDI!
Welcome to Macronutrient Extraction and Determination from Invertebrates! Here's an overview of the extraction protocol:

There are several overnight incubation steps, so don't be alarmed by some of the long procedure times!
Collection and preparation of materials
Collect invertebrates and kill them, ideally by freezing.
Desiccate the invertebrate specimens via freeze-drying or heat-drying until completely dry.
Weigh the dried invertebrate using appropriately sensitive scales, recording the mass.
Determination of exoskeletal mass
For the greatest accuracy, exoskeletal mass determination should be carried out in parallel to the macronutrient analyses with different specimens, but it is possible to streamline these together if necessary. If you do wish to streamline them, the protocol for exoskeletal mass determination largely follows the steps for the macronutrient determination, so attention will be drawn to the overlapping aspects. In order to streamline these processes, exoskeletal determination should take place after extraction of samples for lipid, and carbohydrate and protein determination. When lysing the tissue for protein and carbohydrate determination, the exoskeleton should instead simply be cracked to facilitate penetration of the tissue by the NaOH without breaking apart the exoskeleton itself.
Add 1 ml of 1:12 chloroform:methanol to each sample tube and leave the tube containing the specimen and chloroform:methanol at room temperature for 24 hours.
Discard the supernatant, avoiding the removal of any tissue, add another 1 ml of 1:12 chloroform:methanol to each sample tube and leave the tube containing the specimen and chloroform:methanol at room temperature for 24 hours.
Discard the supernatant, avoiding the removal of any tissue, and evaporate any remaining residual solvent.
Add 0.1 M NaOH to the sample tube, aiming for 5-10X the body volume of the invertebrate. Lightly crack the exoskeleton to expose the inner tissues, and incubate at 80°C for 2 h.
Allow the sample to soak overnight at room temperature.
Discard the supernatant, avoiding the removal of any tissue. Add a further round of 0.1 M NaOH (the same volume) to the sample tube, and incubate at 80°C for 2 h.
Allow the sample to soak overnight at room temperature.
Discard the supernatant, avoiding the removal of any tissue. Add water to the tube (the same volume as with NaOH) and remove (without removing any exoskeletal mass) to wash away any remaining NaOH.
Repeat the water wash in the previous step so that it has been carried out twice in total.
Remove the water (without removing any exoskeletal mass) and evaporate any remaining water.
Weigh the remaining exoskeleton in the tube and subtract the mass of the tube to calculate the exoskeletal mass.
Determination of lipid content
Add 1 ml of 1:12 chloroform:methanol to each sample tube and leave the tube containing the specimen and chloroform:methanol at room temperature for 24 hours.
Remove 500 μl of chloroform:methanol solution and keep it in a separate tube for lipid analysis. Unless the invertebrate dry body mass is >10 mg, the remaining solution in the original tube is taken forward for determination of protein and carbohydrate content (STEP 31), ideally with immediacy given additional overnight steps. If the body mass is >10 mg, the sub-steps below detail gravimetric lipid determination.
From the original tube containing the remaining chloroform:methanol solution and the invertebrate tissue, discard the remaining supernatant (having already removed 500 μl for lipid analysis) avoiding the removal of any tissue. Add another of the same volume of 1:12 chloroform:methanol as in STEP 17 to each sample tube and leave the tube containing the specimen and chloroform:methanol at room temperature for 24 hours.
Discard the supernatant, avoiding the removal of any tissue, and evaporate any remaining residual solvent.
Weigh the remaining body mass and subtract it from the original dry mass to determine total lipid mass gravimetrically.
If possible, estimate the lipid content of the invertebrates and dilute accordingly to take forward a sub-sample of 0.5-1.75 mg/ml.
The lipid content of the 500 μl of chloroform:methanol solution removed during STEP 18 can be determined using the sulfo-phospho-vanillin method.
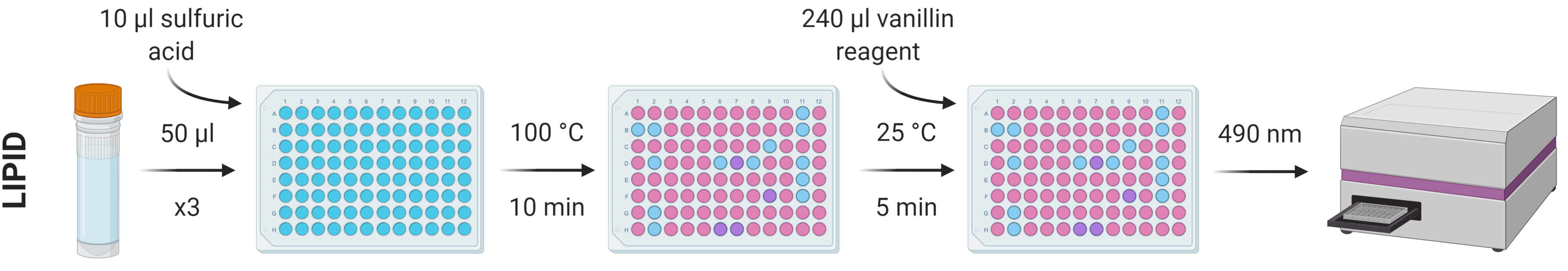
Prepare a stock standard dilution series using a suitable analogue (e.g. analytical lard oil) of known concentration diluted with 1:12 chloroform:methanol. A dilution series of 0-2 mg/ml in nine increments (0, 25, 125, 250, 500, 750, 1000, 1500 and 2000 μg/ml) should suitably cover a good range of concentrations, but this can be adjusted accordingly.
Make up the vanillin reagent using the following amounts (which should account for reagent overage) per sample/standard repeat (so thrice per sample/standard if running triplicates, as advised):
| A | B |
|---|---|
| Reagent | Amount |
| Vanillin | 0.33 mg |
| Hot water | 55 μl |
| 85% phosphoric acid | 220 μl |
From each standard and sample, put three repeats of 50 µl into a flat-bottomed 96-well plate and heat at 100 °C in a ventilated hood until all solvent has evaporated (leaving just the lipid residue; ~10 min).
Add 10 µl concentrated sulfuric acid to the lipid residues and vortex/mix before incubating again at 100 °C for 10 min.
Allow the samples to cool to room temperature and add 240 µl vanillin reagent to each well, vortexing/mixing for homogenous colouration.
After 5 min, transfer 200 µl of each well into a new plate.
Measure absorbance at 490 nm using a spectrophotometer.
Determination of protein and carbohydrate content
From the original tube containing the remaining chloroform:methanol solution and the invertebrate tissue, discard the remaining supernatant (having already removed 500 μl for lipid analysis) avoiding the removal of any tissue. Add another 1 ml of 1:12 chloroform:methanol to each sample tube and leave the tube containing the specimen and chloroform:methanol at room temperature for 24 hours.
Discard the supernatant, avoiding the removal of any tissue, and evaporate any remaining residual solvent.
Lyse the tissue and ensure it is mixed/homogenous. Lysis can be carried out using a pestle and mortar, or bead-beating method, ensuring all available tissue is taken forward (unless the dry body mass exceeds 10 mg, in which case follow the below sub-step).
If working with invertebrates with a dry body mass >10 mg, lyse/homogenise the body and take a 3-7 mg subsample forward for the subsequent steps.
Add 1 ml 0.1 M NaOH to the sample tube. Incubate at 80°C for 30 min in a themo-shaker to ensure mixture of the lysed material with the NaOH.
Allow the sample to soak overnight at room temperature.
Centrifuge the sample for 10 min at 13,000 rpm.
Take 600 µl of the supernatant into a separate tube for taking forward for protein and carbohydrate determination.
If possible, estimate the protein and carbohydrate content of the invertebrates and dilute accordingly for each assay to take forward 0.5-1.75 mg/ml.
The carbohydrate content of the 600 μl of NaOH solution removed during STEP 34 can be determined using the anthrone method.
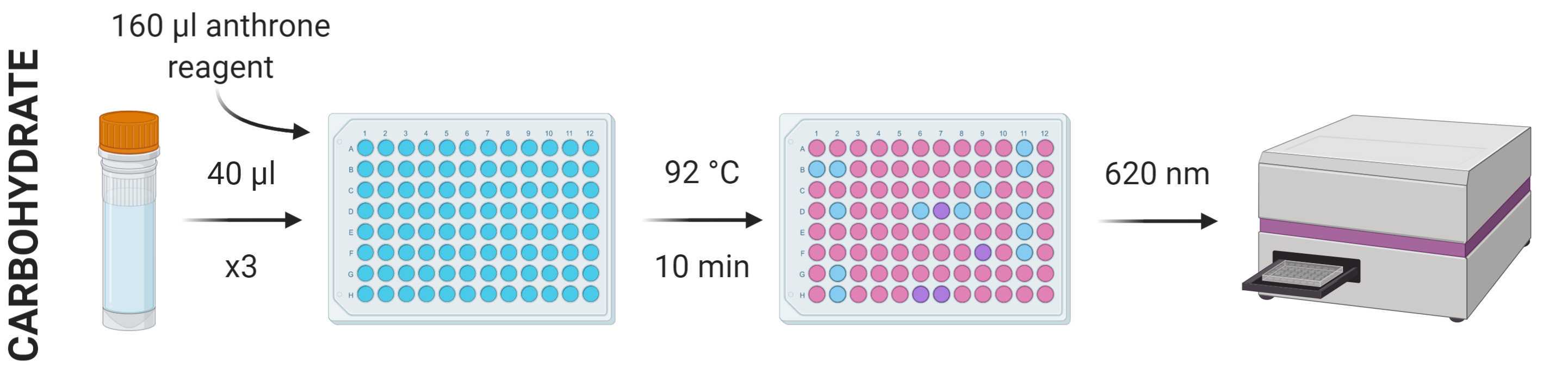
Prepare a stock standard dilution series using a suitable analogue (e.g. analytical corn starch) of known concentration diluted with polished water. A dilution series of 0-2 mg/ml in nine increments (0, 25, 125, 250, 500, 750, 1000, 1500 and 2000 μg/ml) should suitably cover a good range of concentrations, but this can be adjusted accordingly.
Make up the anthrone reagent using the following amounts (which should account for reagent overage) per sample/standard repeat (so thrice per sample/standard if running triplicates, as advised):
| A | B |
|---|---|
| Reagent | Amount |
| Anthrone | 185 mg |
| Concentrated sulfuric acid | 185 μl |
From each standard and sample, put three repeats of 40 µl into a flat-bottomed 96-well plate, add 160 µl anthrone reagent to each and vortex/mix.
Incubate the plate at 92 °C in a ventilated hood for 10 min.
Cool the plate to room temperature and measure absorbance at 620 nm using a spectrophotometer.
The protein content of the 600 μl of NaOH solution removed during STEP 34 can be determined using one of several colorimetric protein methods. The three highlighted in the MEDI manuscript (the benefits and drawbacks also discussed therein) are summarised in the image below. This protocol will describe the protocol for the Lowry assay. The reagents involved in each of these assays are widely commercially available.
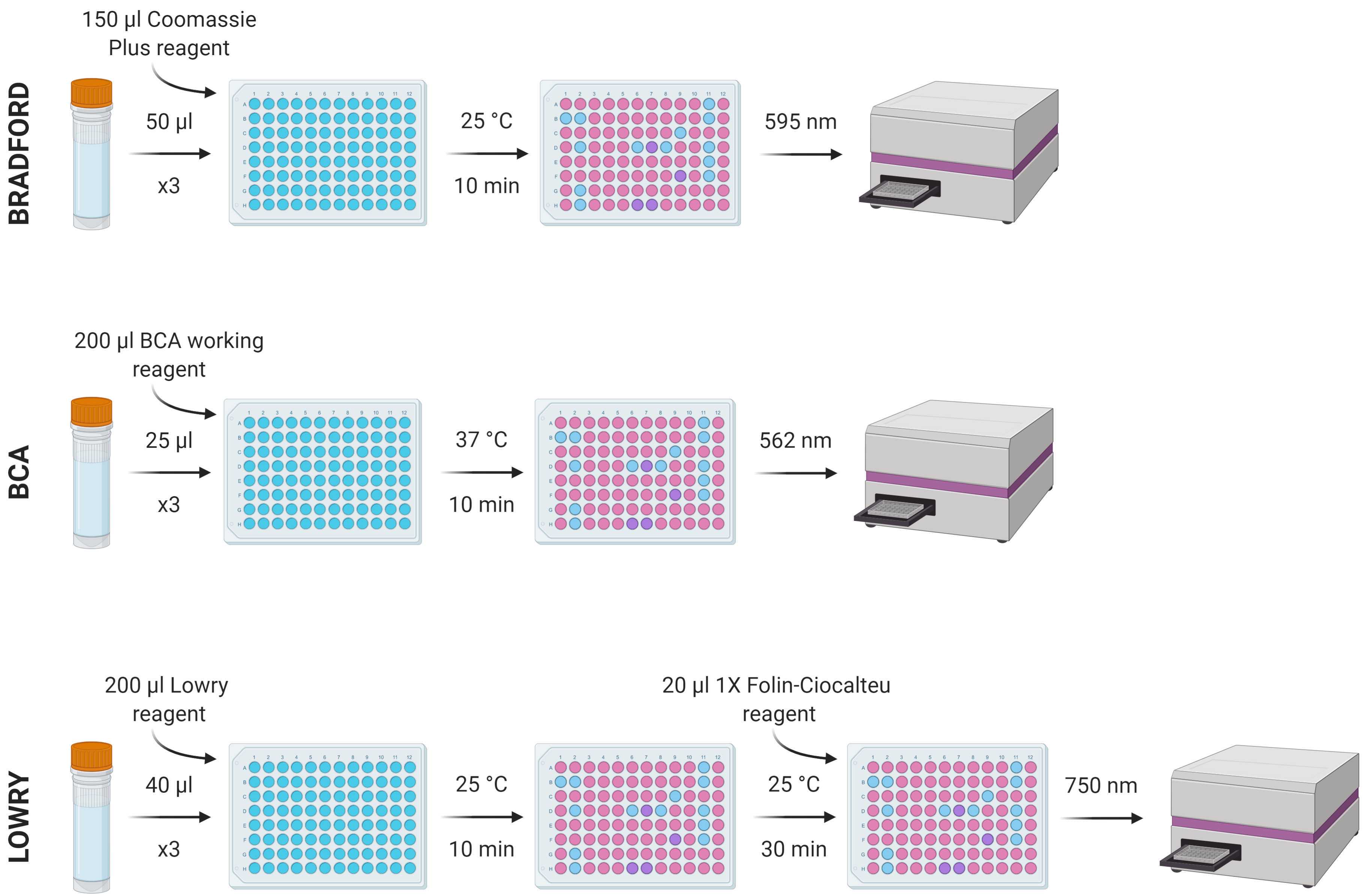
Prepare a stock standard dilution series using a suitable analogue (e.g. bovine serum albumin) of known concentration diluted with polished water. A dilution series of 0-2 mg/ml in nine increments (0, 25, 125, 250, 500, 750, 1000, 1500 and 2000 μg/ml) should suitably cover a good range of concentrations, but this can be adjusted accordingly.
From each standard and sample, put three repeats of 40 µl into a flat-bottomed 96-well plate, add 200 µl of Modified Lowry Reagent to each and vortex/mix.
Incubate at room temperature (~20-25 °C) for 10 min.
Add 20 µl of 1X (1N) Folin-Ciocalteu reagent to each well and vortex/mix.
Incubate at room temperature (~20-25 °C) for 30 min.
Measure absorbance at 750 nm using a spectrophotometer.
