Light-Seq Cell Barcoding
Jocelyn Y. Kishi, Ninning Liu, Emma R. West, Kuanwei Sheng, Jack J. Jordanides, Matthew Serrata, Constance L. Cepko, Sinem K. Saka, Peng Yin
Abstract
We present Light-Seq, an approach for multiplexed spatial indexing of intact biological samples using light-directed DNA barcoding infixed cells and tissues followed by ex situ sequencing. Light-Seq combines spatially targeted, rapid photocrosslinking of DNA barcodes onto complementary DNAs in situ with a one-step DNA stitching reaction to create pooled, spatially indexed sequencing libraries. This light-directed barcoding enables in situ selection of multiple cell populations in intact fixed tissue samples for full-transcriptome sequencing based on location, morphology or protein stains, without cellular dissociation. Applying Light-Seq to mouse retinal sections, we recovered thousands of differentially enriched transcripts from three cellular layers and discovered biomarkers fora very rare neuronal subtype, dopaminergic amacrine cells, from only 4–8 individual cells per section. Light-Seq provides an accessible workflow to combine in situ imaging and protein staining with next generation sequencing of the same cells, leaving the sample intact for further analysis post-sequencing.
Before start
This protocol is for selective barcoding of cells seeded onto an Ibidi 18-well µ-slide. A separate protocol can be found for selective barcoding of tissue samples.
Clean workspace (bench, pipettes, etc.) with 70% ethanol before starting.
When the protocol calls for water, always use UltraPure water.
All reagents should be molecular-grade and RNAse free.
All reactions with enzymes are prepared on ice.
Key Equipment:
Microscope
Light-Seq requires an optical system that can focus UV light onto specific regions of interest. In the publication, we employ a wide field microscope with a 365 nm LED and a digital micromirror device (DMD) to accomplish this, but other systems are also amenable. We have also tested a point-scanning confocal microscope with a 405 nm laser. We recommend consulting your microscopy core or representative to discuss the best solution at your institution.
Flat-top Thermocycler
The first few steps of the protocol require thermal cycling and incubations of tissue slides. For this, we recommend the Eppendorf® Mastercycler® nexus Flat Thermal Cyclers (VWR Cat No. 71003-568).
Steps
Protocol Overview
This protocol is for selective barcoding of cells seeded onto an Ibidi 18-well µ-slide for our Light-Seq paper. https://www.nature.com/articles/s41592-022-01604-1
A separate protocol can be found for selective barcoding of tissue samples.
https://www.protocols.io/view/light-seq-x54v9jno4g3e/v1
The protocol will take roughly one week with some pause points. An example schedule can be:
Day 0: Cell seeding and chamber preparation
Cell seeding is an overnight process, should be performed the day before planned barcoding.
Chamber Preparation
An overnight process, should be performed in parallel with cell seeding
Day 1: Cell fixation, in situ reverse transcription, A-tailing
Cell fixation and permeabilization (10-20 minutes)
Reverse transcription (2.5 hours)
A-tailing (1 hour)
Day 2: Barcoding, cDNA displacement and Cross-junction synthesis
Barcoding (0.5 - 2 hours, depending on number of desired ROI's)
cDNA displacement (1 hour)
Cross-junction synthesis (1-2 hours)
Day 3: qPCR amplification
qPCR (5 hours)
Day 4: Library prep for NGS
Library Preparation for Next-Generation Sequencing (3 hours)
Cell line maintenance
For our cell mixing experiment, we used two different cell lines
- HEK-293-GFP (Human)
- 3T3 (Mouse)
Which must be cultured and maintained separately prior to a cell mixing experiment.
Cell lines
Reagents needed:
HEK293-GFP Cell line growth media
DMEM, GlutaMax growth media supplemented with:
| A | B |
|---|---|
| Reagent | Concentration |
| Fetal Bovine Serum (FBS) | 10% |
| MEM non-essential amino acids | 0.1 mM |
| Penicillin Streptomycin | 1% |
| Blasticidin | 10 μg/ml |
The blasticidin is supplemented into the growth media at in order to act as a selective marker for the eGFP expression cassette inserted into the HEK cells.
For growing the HEK-GFP cells after an initial thaw from a frozen aliquot, it is recommended to grow the cells in a flask with pre-warmed 37 C media without blasticidin for 1 night.
- The blasticidin can be too much of a stressor after the initial thaw and will stunt growth.
The following day, replace the media with pre-warmed 37C growth media with blasticidin.
3T3 Cell line growth media
DMEM, GlutaMax growth media supplemented with:
| A | B |
|---|---|
| Reagent | Concentration |
| Calf Bovine Serum (BCS) | 10% |
| Penicillin Streptomycin | 1% |
Note that the ATCC website recommends their 3T3 cells to be grown in Calf Bovine Serum rather than Fetal Bovine Serum. We have found that the cells do indeed grow far better in the calf serum than the FBS serum.
Cell splitting and subculture protocol
Our cell splitting and subculturing protocol generally follows the recommended ATCC protocols that are listed on their website for all their cell lines:
https://www.atcc.org/products/crl-1658
Reagents needed
Cell Splitting
The following protocol assumes a standard T-75 corning flask for volumes.
Pre-warm all buffers to be used to 37C.
- Aspirate out cell growth media with a pipette. Aspirate away from cells.
- Wash cells with 10 ml of DPBS.
- Aspirate out the DPBS media
- Add 2-3 ml of Trypsin and coat the surface evenly.
- Place flask in the 37C incubator for 5-10 min. Cells should detach in that time.
- Add 7-8 ml of DMEM growth media to inactivate the trypsin.
- Pipette media up and down to de-clump cells. Wash the entire flask surface with media to get any cells that are still adhering to the flask.
Splitting and re-plate the cells.
To do a quick and dirty split without a cell counter, we typically dilute the cells to some volume in fresh media depending on how long we want to wait before the cells become confluent.
A rule of thumb that we use is:
1 ml in 10 ml (1:10) - 1-2 days
0.5 ml in 10 ml (1:20) - 3 days
0.4 ml in 10 ml (1:25) - 4 days
Incubate in a cell culture incubator at 37C and 5% CO2
Preparing the ibidi Chamber (overnight)
While the cells are growing, it is good to start preparing the chamber the day before you plan to seed the cells.
The chamber itself will initially be coated with Poly-L-Lysine (PLL) from the vendor and it will be coated again with Poly-D-Lysine (PDL). We find that a double coating greatly improves cell adhesion, especially for HEK cells. This is important in order for the cells to be robust against the repeated washing steps with 60% formamide.
Reagents needed:
Prepare PDL aliquots
- Dissolve PDL to a working concentration of
0.3mg/mLin 2X Borate buffer (diluted in water) - Store aliquots at
-20°C
Chamber Coating
All steps with the chamber open should be ideally performed in a sterile fume hood to prevent contamination during drying steps
- Take a clean 18-well PLL chamber and pipette 100 μl of the PDL solution into each well
- Place the lid back on the chamber, cover and leave in 4C, overnight .
- In the hood, pipette off the PDL solution and dry in hood for 1 hour .
- Wash each well with UltraPure water ( 100 μl ) X3.
- Dry in hood for 1 hour.
Cell Seeding
Cell seeding and co-culturing
The day before the barcoding experiment, we will then need to seed and co-culture both cell lines in a single ibidi well.
Co-culture growth media
DMEM, GlutaMax growth media supplemented with:
| A | B |
|---|---|
| Reagent | Concentration |
| Calf Bovine Serum (BCS) | 10% |
| MEM non-essential amino acids | 0.1 mM |
| Penicillin Streptomycin | 1% |
The growth media here is a combination of the two different cell growth media. We find that HEK can grow equally well in Calf Bovine Serum.Blasticidin must be removed, since the 3T3 cells do not carry the resistance marker for it.
Detach cells from flask and re-suspend in 1 ml growth media
- Aspirate out media with a pipette (2ml glass) Aspirate away from cells.
- Wash with 10 ml of DPBS (pre-warmed)
- Aspirate out media (2 ml glass)
- Add 2 ml of Trypsin, coat the well evenly. (5 ml pipette)
- Place 2 min in incubator ( Minimize time in trypsin!, check if detached after 2 min)
- Add 8 ml of Co-culture media (w/ BCS), should inactivate rest of trypsin. (10 ml pipette)
- Pipette media up and down, using the surface to remove adherent cells.
- Put media in 10 ml tube and spin down for 5 min at 200 RCF.
- Aspirate cell media and resuspend in 1 ml media.
Measure cells with a cell counter
This step is highly recommended in order to seed the cells to an optimal density in the wells.
Put 10 ul of cell media with 10 ul of dye for cell counter.
Seed Cells
-
Pre-warm the co-culture growth media to 37C.
-
Take the prepped 18-well ibidi chamber and add 80 μl of pre-warmed media to all the wells that you want to seed the cells in.
-
Using cell counter, seed with 10 ul for EACH cell into the 18-well ibidis for a total of 100 μl.
If we want to seed 5k cells, then we need to dilute the cell culture to a density of 500 cells/μl By pipetting a small volume of cells into a larger volume of media, we ensure a more even cell deposition. -
Create a pre-mixture of both cell lines into a stock tube. ~200 μl.
-
Pipette 20 μl into each well that requires the mixture. Pipette up and down to mix.
-
Grow overnight in 37C incubator.
Fixation, Permeabilization, Reverse Transcription
Reagents:
Reagents for Reverse Transcription
| A | B | C |
|---|---|---|
| Reagents | Supplier | Cat. No. |
| Thermo Scientific™ Maxima H Minus Reverse Transcriptase (200 U/μL) (includes 5X buffer) | Thermo Scientific | FEREP0753 |
| Triton-X-100 | Sigma Aldrich | T8787- 50ML |
| 5M NaCl | Invitrogen | AM9760G |
| TWEEN® 20 | Sigma Aldrich | P9416- 50ML |
| PBS | Invitrogen | AM9625 |
| Formamide | Invitrogen | AM9342 |
| UltraPure Water | Invitrogen | 10977023 |
| Deoxynucleotide (dNTP) Solution Mix - 8 μmol at 10mM each | NEB | N0447S |
| RNaseOUT™ Recombinant Ribonuclease Inhibitor | Invitrogen | 10777019 |
Reverse Transcription DNA Oligos
| A | B | C | D | E |
|---|---|---|---|---|
| Name | Description | Sequence | Supplier | Purification |
| RT.5N.3G | RT Primer | TTTACACGATTGAGTTATNNNNNGGG | IDT | HPLC |
Recommended to dilute this primer to a 10 uM stock.
Buffers Fixation buffer (4% FA) Prepare in fume hood if possible, formaldehyde is toxic. * Take one ampule of 16% FA and dilute 1:4 in 1X PBS
- Store aliquots in -20C
Permeabilization buffer (0.25% Triton X-100)
4 ml 1X PBS
10 µl Triton X-100
1X PBS (40 ml)
4 ml 10X PBS
36 ml Ultrapure water
0.1% PBS-Tween (30 ml)
30 ml 1X PBS
30 ul Tween-20
Stringent Wash (0.1% PBS-Tw + 60% formamide):
3 mL 10X PBS
18 mL 100% formamide
9 mL UltraPure H2O
30 μL Tween-20
Note: Note: Store at 4C for use later during barcoding.
High-Salt Wash (1X PBS + 1 M NaCl + 0.1% Tween- 20):
4 mL 10X PBS
8 mL 5 M NaCl
28 mL UltraPure H2O
40 μL Tween-20
Note: Store at room temperature for use later during barcoding.
10% Triton X-100 (100 μl):
10 μL Triton X-100
90 μL UltraPure water
Note: Mix very well by vortexing. Recommended to make this in a 2 mL tube for better mixing.
Reverse Transcription Master Mix (RTMM)
| A | B | C |
|---|---|---|
| Reagent | Reaction concentration | μL reagent per 50μL reaction |
| 5X RT buffer | 1x | 10 |
| 10 mM dNTPs | 300 μM | 1.5 |
| UltraPure water | 26 | |
| 10% Triton X-100 | 0.5% | 2.5 |
| 10 μM RT.5N.3G primer | 1 μM | 5 |
| 100 mM RnaseOUT | 6 mM | 3 |
| Maxima RT H Minus (200 U/uL) | 8 U/μL | 2 |
| Total | 50 |
Note: Keep on ice. Leave out RnasaOUT and Maxima RT H Minus until final step.
Fixation and permeabilization
- Remove ibidi chamber with cells from the incubator and carefully remove the cell media. Cells may not be super adherent prior to fixation.
- Wash once with pre-warmed pre-warmed 37C DBPS.
- Warm 4% FA to room temperature. Fix cells by adding ~75 μl of 4% FA into each well for 10 minutes at room temperature.
- Wash wells 2x with 1X PBS.
- Permeabilize cells with 0.25% Triton for 10 minutes at room temperature.
- Wash wells 2x with 1X PBS.
- Immediately move to reverse transcription.
In situ In situ Reverse transcription.
Add enzymes (Maxima RT H minus and RNAse Out) to Reverse Transcription Master Mix (RTMM) and mix well.
-
After washing wells with 1XPBS, add 50ul of RTMM to each well.
-
Place slide on a flat-top thermocycler.
Reverse Transcription Thermocycler Program
12-cycle ramp program (Lid: 60°C):
Phase 1:
22°C 30 mins
Phase 2 (12 cycles):
8°C 30 s
15°C 30 s
25°C 30 s
30°C 1 min
37°C 1 min
42°C 2 min
Phase 3:
42°C 30 min
4°C Forever
3. Wash each well 3 x 5 min in Stringent Wash.
-
Wash each well 2 x 2 min in High Salt Wash.
-
Wash each well 2 x 2 min in 0.1% PBS-Tw.
-
Resuspend in 0.1% PBS-Tw.
Optional Pause Point : Store at 4C overnight before A-tailing.
A-Tailing
Reagents needed:
We are only using the dATP from the dNTP set
Prepare Aliquots
-
Create aliquots of 10 mM dATP (20 µL aliquots) and 25 mM ddATP (~3 µL aliquots). Store at -20C.
Both reagents are supplied in ultrapure water. -
Dilute 25 mM ddATP stock further to 250 μM (1:100 dilution from 25 mM stock).
Buffers A-tailing master mix
| A | B | C | D |
|---|---|---|---|
| Reagent stock concentration | Reaction concentration | uL reagent per 50uL reaction | Cat no. |
| 10X ThermoPol Reaction Buffer | 1X | 5 | B9004S |
| 10 mM dATP | 1 mM | 5 | N0446S |
| 250 μM ddATP | 25 μM | 5 | GE27-2051-01 |
| Ultrapure water | 32.5 | 10977023 | |
| TdT enzyme (20,000U/mL) | 1000 U/mL | 2.5 | M0315L |
| Total | 50 |
0.1% PBS-Tw (30 mL):
30 mL 1X PBS
30 μL Tween-20
A-tailing protocol 1. Wash wells once (from previous step) with 0.1% PBS-Tw.
- Aspirate, add 50 μL A-tailing Master Mix and incubate for 30 min at 37C .
- Wash 3 x 1min in 0.1% PBS-Tw.
Optional Pause Point: Store at 4C overnight before barcoding.
Light-Directed Barcoding of cDNAs
Light-directed carcoding of select Cells for sequencing
Light-directed barcoding is performed sequentially for different regions of interest/cell populations within the same sample. For the cellmixing experiment, we barcoded roughly 25 cells for each cell line for a total of 50 cells per well.
Light-Seq barcodes used
In the paper, we barcoded the HEK cells first with barcode 1 (Cy5).
Then 3T3 cells second with barcode 2 (Cy3).
| A | B | C | D | E |
|---|---|---|---|---|
| GATE.D12.B1 | Barcode sequence 1 - Cy5 labeled barcode strand. | GGAGTTGGAGTGAGTGGATGAGTGATGDDDDDDDDDDDDTATGGATGAGTTATATAACTCA[cnvK]TCGTGTAAAT[Cy5-3] | GeneLink | PAGE |
| GATE.D12.B2 | Barcode sequence 2 - Cy3 labeled barcode strand. | GGAGTTGGAGTGAGTGGATGAGTGATGDDDDDDDDDDDDGTTAGGTGAGTTATATAACTCA[cnvK]TCGTGTAAAT[Cy3-3] | GeneLink | PAGE |
Note: Fluorescently labelled barcode strands are light-sensitive and should be protected from light if left out for long periods of time, although some ambient light is acceptable. Ambient light is generally far too low power to excite a fluorophore or activate the crosslinker. We routinely do all steps in well-lit rooms, but cover the samples and tubes with foil during incubation periods.
Barcoding Reagents
| A | B | C |
|---|---|---|
| Reagents | Supplier | Cat. No. |
| Dextran Sulfate 50% Solution | Sigma Aldrich | S4030 |
| Salmon-sperm DNA | Thermo Fisher | AM9680 |
| 5 M NaCl | Invitrogen | AM9760G |
| TWEEN® 20 | Sigma Aldrich | P9416- 50ML |
| PBS | Invitrogen | AM9625 |
| Formamide | Invitrogen | AM9342 |
| UltraPure Water | Invitrogen | 10977023 |
| Barcode DNA Oligos (see sequences table) | GeneLink |
From the buffers we prepared in Step 5. We will also need.
1X PBS (40 ml)
4 ml 10X PBS
36 ml Ultrapure water
0.1% PBS-Tween (30 ml)
30 ml 1X PBS
30 ul Tween-20
Stringent Wash (0.1% PBS-Tw + 60% formamide):
3 mL 10X PBS
18 mL 100% formamide
9 mL UltraPure H2O
30 μL Tween-20
Note: Note: Store at 4C for use later during barcoding.
High-Salt Wash (1X PBS + 1 M NaCl + 0.1% Tween- 20):
4 mL 10X PBS
8 mL 5 M NaCl
28 mL UltraPure H2O
40 μL Tween-20
Note: Store at room temperature for use later during barcoding.
Barcode Hybridization Solution (See note at top of section)
| A | B | C |
|---|---|---|
| Reagent | Reaction concentration | μL per 50 μL well** |
| 10X PBS | 1x | 5 |
| 5M NaCl | 500 mM | 5 |
| UltraPure water | 38.7 | |
| TWEEN® 20 | 0.1% | 0.05 |
| Total | 48.75 |
**Dextran sulfate makes this mix viscous, so it is recommended to make significant excess (~1.2X what is needed for the number of wells).
Barcoding Solution (250 nM final concentration)
Barcode 1 Solution (per 50 uL well - make excess!):
1.25 μL of 10uM Barcode Strand (GATE.D12.B1)
48.75 μL hybridization master mix
Barcode 2 Solution (per 50 uL well - make excess!):
1.25 μL of 10uM Barcode Strand (GATE.D12.B2)
48.75 μL hybridization master mix
Barcoding Workflow
-
Wash 1x with High Salt Wash.
-
Aspirate out High-salt and add 50 ul of Barcode 1 solution.
Incubate for **30 minutes** at room temperature. Cover from light. -
Aspirate to remove all liquid, then wash 3 x 1 min in High-Salt Wash.
This removes most of the un-hybridized barcoding strands. The barcoding strand has enough base-pairing to be relatively stable in high-salt buffer. -
Sample is now ready to be barcoded, bring to instrument and draw ROI's around desired regions.
See supplemental in publication for proper alignment of the photomask. -
Wash wells 2x with Stringent Wash . Let sit in stringent wash for 2 minutes on last wash.
Repeat this step three more times, for a total of 4 x 2 = **8 washes.** -
Wash 2 x 2 min in High-Salt Wash , then add fresh High-Salt Wash.
-
Repeat steps 1-6 for Barcode 2.
Transfer to 0.1% PBS-Tween and optionally image barcoded cDNAs to visualize.
Optional Pause Point: Store at 4C overnight in a humidified chamber.
Displacement of cDNAs and cross-junction synthesis
Now that cDNAs are barcoded and A-tailed, they must be extracted for library preparation and sequencing. To do this, the RNA template is digested by mild RNaseH treatment, which degrades the RNA bound to the barcoded cDNAs in situ, liberating the barcoded cDNAs for collection.
The workflow for cDNA displacement is largely similar to the one we used for the retina tissue.
Note: Use low retention tips.
Displacement Reagents
| A | B | C |
|---|---|---|
| Reagents | Supplier | Cat. No. |
| ThermoPol® Reaction Buffer Pack | NEB | B9004S |
| RNase H - 1,250 units | NEB | M0297L |
| UltraPure Water | Invitrogen | 10977023 |
Cross-Junction Synthesis Reagents
| A | B | C |
|---|---|---|
| Reagents | Supplier | Cat. No. |
| Bst DNA Polymerase, Large Fragment- 8,000 units | NEB | M0275L |
| ThermoPol® Reaction Buffer Pack | NEB | B9004S |
| UltraPure Water | Invitrogen | 10977023 |
| Deoxynucleotide (dNTP) Solution Mix - 8 μmol at 10mM each | NEB | N0447S |
Cross-Junction Synthesis Primer r
| A | B | C | D | E |
|---|---|---|---|---|
| Name | Description | Sequence | Supplier | Purification |
| GATC.20T.p | Primer for Cross-Junction Synthesis | GAGAATGTGAGTGAAGATGTATGGTGATTTTTTTTTTTTTTTTTTTT | IDT | HPLC |
Buffers Displacement Mix
| A | B | C |
|---|---|---|
| Reagent | Reaction concentration | uL reagent per 50 uL well |
| 10x ThermoPol Reaction Buffer | 1x | 5 μL |
| RNase H (from NEB) (5,000U/mL) | 250 U/mL | 2.5 μL |
| UltraPure Water | 42.5 µL | |
| Total | 50 |
Displacement Workflow
-
Aspirate out buffer from previous step and add 67.5 μl of Displacement Mix to each well.
Incubate on flat-top incubator for **37C for 30 minutes.** -
Prepare a set of lo-bind PCR tubes that will collect the eluate from each well by pipetting 1.6 μl of 1μM GATC.20T.p cross-junction synthesis primer into each tube.
Pre-pipetting the primer into the tube will minimize the number of pipette steps the cDNA library is subjected to. For example, adding the primer after collection will introduce another pipette tip into the solution which some cDNA's may stick to. -
Prepare a set of 20 nM GATC.20T.p primer solution. Take a low-retention tip and pipette this solution up and down and three times to "pre-block" it with the primer DNA.
Pipetting this solution with a tip helps to block the tip with some DNA to prevent any cDNA from sticking to the tip surface. Take care not to generate bubbles during this step. -
With the same empty tip from step 3, move to a well and carefully pipette the solution in the well up and down three times.
The well should still be in displacement mix along with the now displaced cDNA's. It is important to not over-pipette or introduce bubbles into the solution at this stage. The mild agitation we introduce with pipetting up and down should help resuspend the cDNA library. -
Collect the eluate and place in a collection tube that contains the GATC.20T.p primer. Pipette up and down a few times to mix the primer.
-
Repeat steps 1-5 with a fresh tip for all wells.
-
Heat inactivate the collection tubes in 75C for 20 min.
-
Re-hydrate the chamber wells and wash wells 3 times with 0.1% PBS-Tw.
The chamber containing the samples can be stored for follow-up immunostains. -
Move on to Cross-junction synthesis.
Optional: Samples can be imaged after displacement to verify that the fluorescent barcode signal is no longer present. At this point, follow-up stains can be performed on the sample including antibody stains, H&E, etc.
Barcoded sample
 and 3T3 cells (mouse)")
Displaced samples

Cross-junction synthesis
- Add 10.9 μL Cross-Junction Synthesis Mix to each tube:
Cross-Junction Synthesis Mix
| A | B | C |
|---|---|---|
| Reagent | Final Reaction concentration | uL reagent per 80 uL well |
| 10X ThermoPol buffer | 1x | 1.25 |
| 10mM dNTPs | 100 μM | 0.8 |
| Ultrapure water | 0.85 | |
| Bst LF polymerase (8,000U/mL) | 800U/mL | 8 |
| Total | 10.9 |
Note: These volumes are for 80 uL wells, and volumes should be scaled according to well volume. Note that the primer (GATC.20T.p) was added in the previous step, before heat inactivation.
-
Vortex and spin briefly.
-
Incubate in a thermocycler for:
37C for 30 mins 80C for 20 mins (heat inactivation) -
The resulting products are CJS Samples . Proceed to qPCR to amplify CJS Samples.
Optional Pause Point : Store at -20C overnight.
qPCR Amplification
Cross-Junction Synthesis libraries of barcoded cDNAs are now amplified and prepared for sequencing. We tested both the Kapa Hifi HotStart and Taq polymerase for this step and saw similar results for both. Only the Kapa Hifi results were published in the paper.
We recommend performing qPCR with 30 cycles on a small subset of each sample (5 uL sample in a 10 uL PCR), to identify the appropriate cycle number where the yield displays an inflection point for optimal amplification.
qPCR Primers
| A | B | C | D | E |
|---|---|---|---|---|
| Name | Description | Sequence | Supplier | Purification |
| GATE | PCR Primer 1 | GGAGTTGGAGTGAGTGGATGAGTGATG | IDT | HPLC |
| GATC | PCR Primer 2 | GAGAATGTGAGTGAAGATGTATGGTGA | IDT | HPLC |
Reagents for qPCR
| A | B | C |
|---|---|---|
| Reagents | Supplier | Cat. No. |
| SYBR™ Green I Nucleic Acid Gel Stain - 10,000X concentrate in DMSO | Invitrogen | S7563 |
| HiFi HotStart DNA Polymerase, KapaBiosystems | Roche | KK2502 |
| UltraPure Water | Invitrogen | 10977023 |
qPCR to Determine Amplification Cycle Number
-
Vortex CJS Samples.
-
Dilute Sybr Green I dye in water to make 5x solution (e.g. dilute 10,000X stock twice, 1:100 in water, then 1:20 in water).
-
Create PCR master mix:
| A | B |
|---|---|
| Kapa qPCR Master Mix | |
| Reagent | µL reagent per 10 µL reaction |
| 5x SYBR Green I | 1 |
| Kapa HiFi HotStart buffer (5x) | 2 |
| 10 µM GATE primer | 0.3 |
| 10 µM GATC primer | 0.3 |
| 10 mM dNTPs from Kapa kit | 0.3 |
| UltraPure water | 0.9 |
| Kapa HiFi Hot Start polymerase | 0.2 |
| Total | 5 |
The ratio of Kapa qPCR Master Mix to Cross-Junction Synthesis Solution is 1:1, so each sample will have a 10 uL total PCR with 5 uL of Kapa qPCR Master Mix and 5 uL of Cross-Junction Synthesis Solution.
-
Quickly vortex and spin down master mix, then add 5 uL master mix into each tube.
-
Add 5 uL of appropriate sample from Cross-Junction Synthesis reaction to each tube.
-
Quickly vortex and spin down reactions.
-
Place into qPCR machine for 30 cycles and run the qPCR Machine Protocol.
qPCR Machine Protocol
98C for 3 minutes
_30 cycles_ **30 cycles of:**
98C for 20 seconds
60C for 30 seconds
72C for 2 minutes
Plate read
72C for 5 minutes
Melting curve analysis
Hold at 4C
8. Check amplification graph to choose appropriate cycle number.
After the test qPCR on a small amount of sample, a PCR is performed on all of the CJS Samples to amplify the entire sample for library preparation and sequencing. The cycle number for amplification is chosen based on the test qPCR in the previous step, to prevent over-amplification. Each Cross-Junction Synthesis product (1 per sample well) is amplified in a separate tube. This protocol is identical to the prior qPCR, but scaled to amplify the entire sample and with a reduced number of cycles.
All of the sample should be amplified. However, some qPCR machines have a limit of 50 uL/reaction. Therefore, the full volume should be split into multiple PCRs to ensure accurate amplification.
Amplification of Full Samples
-
Briefly vortex and spin CJS Samples.
-
Measure the volume of CJS sample in each tube and use to calculate volume of qPCR master mix needed for each sample (1:1 ratio of qPCR master mix : CJS sample).
Note: Ensure that PCR volumes do not exceed the limits for your machine and otherwise, split into multiple tubes.
-
Dilute Sybr Green I dye in water to make 5x solution (e.g. dilute 10,000X stock twice, 1:100 in water, then 1:20 in water).
-
Create qPCR master mix:
| A | B |
|---|---|
| Kapa Full qPCR Master Mix | |
| Reagent | µL reagent per 50 µL reaction |
| 5x SYBR Green I | 10 |
| Kapa HiFi HotStart buffer (5x) | 20 |
| 10 µM GATE primer | 3 |
| 10 µM GATC primer | 3 |
| 10 mM dNTPs from Kapa kit | 3 |
| Ultrapure water | 9 |
| Kapa HiFi hot start polymerase | 2 |
| Total | 50 |
-
Add the entire remaining sample with equal parts master mix to each tube.
-
Quickly vortex and spin down reactions.
-
Place into qPCR machine for exactly XX cycles and run the Full qPCR Machine Thermocycler Program.
Full qPCR Machine Thermocycler Program
98C for 3 minutes
**XX cycles** of:
98C for 20 seconds
60C for 30 seconds
72C for 2 minutes
Plate read
72C for 5 minutes
Hold at 4C
*XX is based on the amplification curves from the test qPCR. This number will likely range between 16 and 25.
- The resulting products are Amplified Samples. Store at -20C.
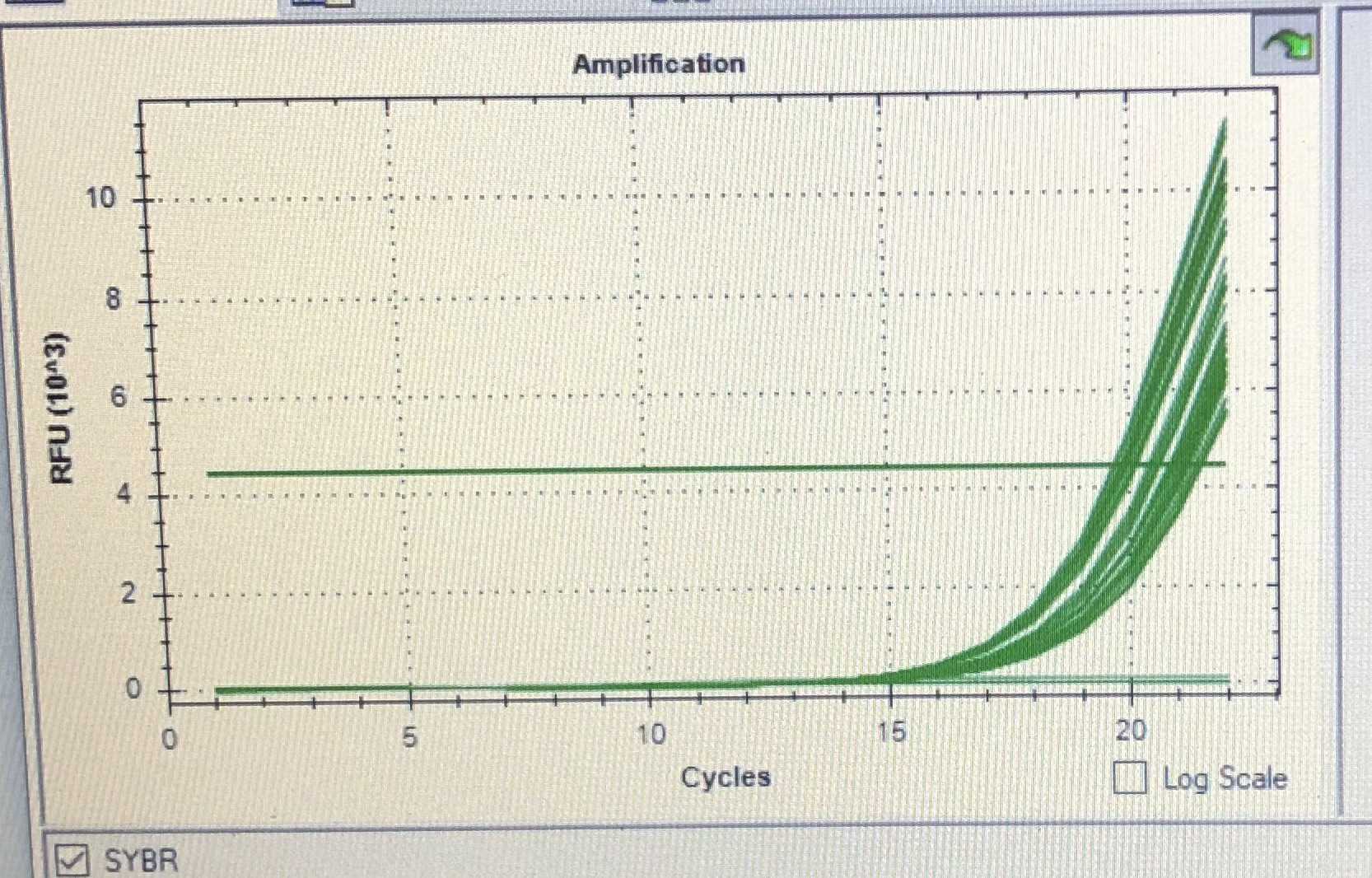
Ideally the amplification will stop right at the exponential phase of the signal.
An example is shown in a phone screenshot below where the amplification is stopped at cycle 22. Past this cycle would be the inflection point and the signal increase will start to slow and eventually plateau. There is generally little benefit to amplifying the library past this point and can also lead to over-amplification of the library.
Example qPCR curve
Library Preparation for Illumina Sequencing
Light-Seq uses conventional tagmentation-based library preparation for Illumina sequencing, but with custom primers for the secondary PCR and for Read 1/i5 sequencing. The custom primers are necessary to specifically enrich for and sequence amplicons containing the light-directed barcode sequences. The first step is bead purification of the amplified libraries for each well, followed by tagmentation, secondary PCR amplification, and a second bead purification.
We generally like to use the magnetic stand rack from permagen
Equipment
| Value | Label |
|---|---|
| 0.2 mL PCR Strip Magnetic Stand | NAME |
| Permagen | BRAND |
| MSR812 | SKU |
Library Preparation Reagents
| A | B | C |
|---|---|---|
| Reagents | Supplier | Cat. No. |
| Magnetic Separator (or equivalent) | Permagen | MSR812 |
| Ampure XP Beads | Beckman | A63881 |
| Ethyl alcohol, pure (200 proof) | Sigma Aldrich | E7023-1L |
| Qubit™ 1X dsDNA High Sensitivity (HS) and Broad Range (BR) Assay Kits | Invitrogen | Q33231 |
| Nextera XT Library Preparation Kit | Illumina | FC-131-1096 |
| UltraPure Water | Invitrogen | 10977023 |
Library Preparation DNA Oligos
These primer sequences are used for unique indexing of samples for pooled sequencing. For each sample, a unique pair of S50X and Next.N70X are required. You do not need to order all primers, only enough pairs to uniquely index your samples of interest. Because the custom i5 index primer does not work well on all Illumina machines, we highly recommend each sample be prepared with a unique i7 index. We hope to adjust our recommendations on this front soon, so stay tuned for updated protocols.
The full table of index primers used can be found in the supplement of our publication
https://www.nature.com/articles/s41592-022-01604-1
as well as the partner protocols.io for the retina
https://www.protocols.io/view/light-seq-x54v9jno4g3e/v1?step=12
Bead purification of Amplified Samples
NOTE: Each well/replicate is processed separately.
The ampure bead purification is based off of the ratio of sample volume to beads. The ampure bead solution contains PEG. The premise is that the the PEG will act as a crowding agent to induce DNA binding to the bead surface, and the concentration of PEG can be adjusted to size select for the desired DNA size. We use a 1.2x ratio for bead purification which should get rid of most DNA <100 bp, <100bp should get rid of most primer dimers and fragmented DNA.
- Make 50 mL of 80% Ethanol, diluted in UltraPure water. (Over time the concentration will slowly decrease but even a 70% Ethanol mixture will also work).
- For each well, combine 80 µL Amplified Sample with 96 µL Ampure XP Beads in a new PCR tube. 1.2x ratio.
- Mix well by pipetting (incubate at room temperature for 5 min ). DO NOT overextend this step.
- Place on Magnetic Separator Stand for 2 min.
- Discard supernatant. Wash 3 x 30 sec with 200 µL of 80% Ethanol.
- Discard ethanol and aspirate remainder with a small 10 µL tip.
- Air dry for ~5 min , until beads are dry. Beads will change color slightly when dry. Try not to overdry the beads, otherwise they will crack.
- Add 30 µL of Ultrapure water to resuspend the beads.
- Remove from the magnetic stand. Mix well by pipetting or vortexing and incubate for 2 min at room temperature.
- Place Magnetic Separator Stand for 1 min. Collect supernatants in new PCR tubes. These are Purified Amplified Samples.
Measure DNA concentrations of Amplified Samples with Qubit HS dsDNA assay.
It is critical that an accurate dsDNA concentration is obtained for the next step. A nanodrop is not sufficient for this purpose. Therefore a Qubit high sensitivity dsDNA assay must be performed on all samples.
This should be done according to the manufacturer's instructions, found here: https://assets.fishersci.com/TFS-Assets/LSG/manuals/MAN0017455_Qubit_1X_dsDNA_HS_Assay_Kit_UG.pdf.
-
Measure concentrations using Qubit Fluorometer.
-
Record concentrations of each Purified Amplified Sample.
Perform Nextera tagmentation
Reagents in this section are from the Nextera XT Library Preparation Kit (Illumina cat. no. FC-131-1096). Tagmentation will fragment the amplified PCR products into shorter sequences, and the length of the reaction time determines how much fragmentation occurs. Therefore, it is very important to follow the times strictly.
-
In a clean set of PCR tubes, aliquot 7 µL of Neutralize Tagment Buffer (NT) . This will be used to stop the tagmentation and prevent over-tagmentation. One tube per sample/well is required. Set aside, next to the thermocycler.
-
To a new PCR tube for each Purified Amplified Sample (on ice):
-
Add 10 µL of Tagment DNA buffer (TD)
-
Add 2 ng of purified Purified Amplified Samples and add water up to 5uL. Calculated sample and water volume to add based on the Qubit concentrations from previous step.
-
Add 5 µL of Amplicon Tagment Mix (ATM) to lid of tube. This mixture contains the tagmentation enzyme. We will spin the tube down to add all enzymes at the same time.
-
-
Briefly spin down tubes, vortex, and spin down. The tagmentation enzyme has now been added.
-
Place into PCR machine and incubate at 55°C for EXACTLY 5 minutes.
-
Immediately stop reactions with 5 µL of Neutralize Tagment buffer (NT).
(Use a multichannel to pipette the buffer from the previously aliquoted PCR tubes in step 1 to mix quickly).
- Vortex and spin to ensure full mixing. Hold Tagmented Samples on ice.
Sample indexing PCR with unique i5 and i7 primer pairs
To pool Tagmented Samples from each well/replicate for sequencing, each sample is assigned a unique pair of i7 and i5 primer sequences. The primer pairings should be decided and documented. Each Tagmented Sample will be amplified briefly in this step to attached the sample-specific i5 and i7 sequences.
- Assign and record unique index primer pairs to each sample. For example:
| A | B | C |
|---|---|---|
| Sample | i5 primer | i7 primer |
| Tagmented Sample 1 (Well 1) | S502.GATE | Next.N701 |
| Tagmented Sample 2 (Well 2) | S503.GATE | Next.N702 |
| Tagmented Sample 3 (Well 3) | S505.GATE | Next.N703 |
| Tagmented Sample 4 (Well 4) | S506.GATE | Next.N704 |
Each sample will receive a different pair of primers in the indexing PCR.
-
To each Tagmented Sample tube from the previous tagmentation step:
-
Add 6.5 µL water
-
Add 1.75 µL of standard Nextera (i7: Next.N70X ) primer (from 10 uM stock)
-
Add 1.75 µL of custom Nextera (GATE, i5: S50X.GATE ) primer (from 10 uM stock)
-
Add 15 µL PCR mix (NPM PCR master mix)
-
-
Vortex and spin down reactions.
-
Place in thermocycler and start Indexing PCR Program.
**Indexing PCR Program** 72°C for 3 minutes 95°C for 15 seconds _12 cycles_ **12 cycles of:** 95°C for 15 seconds 55°C for 15 seconds 72°C for 40 seconds 72°C for 1 minute Hold at 10°C -
The resulting tubes are the Indexed Samples.
Purify indexed samples with Ampure Beads
The final bead purification will remove primers and other reagents from the sample prior to sending off for NGS. We now use a ratio of 0.9x as the indexed samples of interest should be a bit longer now.
- Make 50 mL of 80% Ethanol, diluted in UltraPure water.
- For each well, combine 50 µL of Indexed Sample and mix with 45 µL Ampure XP Beads in a new PCR tube.
- Mix well by pipetting (incubate at room temperature for 5 min ). DO NOT overextend this step.
- Place on Magnetic Separator Stand for 2 min.
- Discard supernatant. Wash 3 x 30 sec with 200 µL of 80% Ethanol.
- Discard ethanol and aspirate remainder with a small 10 µL tip.
- Air dry for ~5 min , until beads are dry. Beads will change color slightly when dry.
- Add 30 µL of water to resuspend the beads.
- Remove from the magnetic stand. Mix well by pipetting or vortexing and incubate for 2 min at room temperature.
- Place on Magnetic Separator Stand for 1 min.
- Collect supernatants in new tubes. These are the Purified Indexed Samples.
Measure DNA concentrations and length of Indexed Samples
-
Measure concentrations using Qubit HS dsDNA assay. This should be done according to the manufacturer's instructions, found here: https://assets.fishersci.com/TFS-Assets/LSG/manuals/MAN0017455_Qubit_1X_dsDNA_HS_Assay_Kit_UG.pdf.
-
Record concentrations of each Purified Indexed Sample.
-
Run 2 uL of each Purified Indexed Sample on a 1% agarose gel.
Sequencing
This library preparation is compatible with standard Illumina next-generation sequencing. Note that custom Read 1 and i5 index primers are required and are listed in the table below.
DNA Oligos for Sequencing
| A | B | C | D |
|---|---|---|---|
| Description | Sequence | Supplier | Purification |
| Custom Read 1 Primer -required for sequencing of amplicons. | CGCCGGAGTTGGAGTGAGTGGATGAGTGATG | IDT | HPLC |
| Custom i5 index primer -required for some Illumina sequencers (see caption). | CATCACTCATCCACTCACTCCAACTCCGGCG | IDT | HPLC |
We note that the custom index primer is compatible with HiSeq, but for NovaSeq, unique i7 indices were needed for de-convolution due to inefficient i5 index sequencing.
We have found adding 60% above the standard concentration of each custom primer works well on many of the Illumina sequencers and would recommend this as a starting point.
Pooling samples and prepping for sequencing.
When preparing a pooled sample for sequencing, the total amount of DNA should not exceed the recommended amount for the NGS platform you are using. For example MiSeq and other instruments typically recommend 4 nM of DNA.
Below is an example prep we used for a MiSeq machine with a 30% PhiX spike in.
-
Prepare freshly diluted 0.2N NaOH.
-
Dilute PhiX to 4nM: 1uL 10nM PhiX stock + 1.5uL water
-
Prepare a pooled sample at 4 nM concentration. The volume needed should be calculated based on the final Qubit reading and the size of the amplicon from the gel.
-
Mix 1.5uL 4nM PhiX + 3.5uL 4nM pool (30% spike in)
-
Add 5uL diluted NaOH , incubate for 5 minutes.
-
Add 1,657 uL Hyb buffer (HT1) , vortex to make 12pM. (HT1 will come with the MiSeq kit).
-
Add 600uL of this sample to Well 17, tape over to avoid dust. This is the sample well.
-
Add 3.2uL P5.GATE.p (Custom Read 1 Primer) into reservoir 12 (use syringe to pull out, spike primer, mix well, replace)
We have had good success with elevated custom primer concentrations on Illumina MiSeq and NextSeq machines. The standard custom primer concentrations worked for HiSeq. For NovaSeq, we haven't seen as good efficiency even with the elevated custom primer concentrations, particularly for the custom i5 index sequencing but hope to have updated recommendations soon. For now, we recommend having unique i7 index sequences for each sample to aid in sequence de-multiplexing, and that you check back here periodically or reach out for updated sequencer-specific recommendations.
Immunostaining
A final immunostaining step can be performed on the cells.
All antibodies were spun down at 10,000 g for 10 min at 4 °C before use. Primary antibody mix (1x) x)
(150 ul) 1x PBS, 0.3% Triton X-100, 5% BSA mixture
(2 ul) Lamin B goat primary
(2 ul) TFAM mouse primary
(2 ul) alpha-tubulin rat primary
Secondary antibody mix (1x)
(150 ul) 1x PBS, 0.3% Triton X-100, 5% BSA mixture
(1 ul) secondary alexa 647 anti-mouse
(1 ul) secondary Cy3b anti-goat
(1 ul) secondary alexa 488 anti-rat
-
Aspirate, add primary antibody solution and incubate at Room temp for 1 hour.
-
Wash 2 x 1 minute with 1xPBS
-
Aspirate, add secondary antibody solution and incubate at Room temp for 1 hour.
-
Wash 2 x 1 minute with 1xPBS
-
Aspirate, add DAPI solution and incubate at Room temp for 5 minutes.
-
Wash 2 x 1 minute with 1xPBS and resuspend in fresh 1xPBS
-
Image
