Guidance for populating GenomeTrakr metadata templates (BioSample and SRA)
Ruth Timme, Errol Strain, Maria Balkey, Tina Lusk Pfefer
Disclaimer
Please note that this protocol is public domain, which supersedes the CC-BY license default used by protocols.io.
Abstract
PURPOSE: Guidance on how to populate NCBI's metadata packages, maximizing interoperability for foodborne pathogen surveillance.
SCOPE : This protocol provides detailed instructions for populating the following two templates:
-
BioSample metadata : guidelines for obtaining and populating metadata templates describing the sample.
-
SRA metadata: Guidelines for populating sequence-level metadata template.
Versions:
v6: Added the One Health Enteric package presented at IAFP 2021 meeting.
v7: Updated the picklists in the GenomeTrakr-extended pathogen package, "GT-pathogen package-OHE v0.2.2.xlsx" and added an incremental update file for the DRAFT One Health Enteric Package that includes extensive edits compared to v6 .
v8: Updated the picklists in the GenomeTrakr-extended pathogen package, "GT-pathogen package-OHE v0.2.2.xlsx". Also provided a direct link to the newly published One Health Enteric package.
v9: Bug fix
v10 : updates to the GenomeTrakr-extended pathogen biosample template ( GT-pathogen package-OHE v0.3.xlsx ) and release of newly available One Health Enteric package custom templates.
Before start
Before collecting sequence data for your isolates, ensure that you can provide the minimum metadata recommended by your coordinating surveilliance body. The INSDC, in collaboration with the Global Microbial Identifer (GMI) (https://www.globalmicrobialidentifier.org), recommends using the Pathogen metadata template for pathogen surveilliance submissions: (NCBI: https://www.ncbi.nlm.nih.gov/pathogens/submit-data/and EMBL-EBI: https://www.ebi.ac.uk/ena/submit/pathogen-data).
Steps
Overview
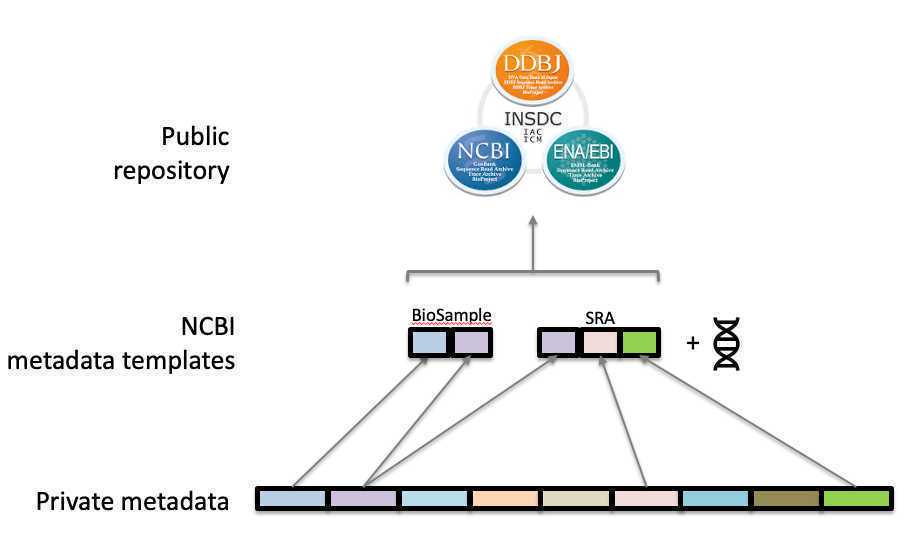
Guidance for organizing and populating the metadata templates required for direct submission to NCBI. This guidance is applicable for most enterics and/or microbial pathogens.
Two metadata templates are required for each NCBI submission:
-
BioSample metadata (metadata describing the sample source and submitter)
-
SRA metadata (metadata describing the sequence data collection)
BioSample metadata template
Templates for BioSample submission:
Laboratories can choose one of the two following templates, offered in Step 2.1 or Step 2.2 .
Validation: Download and populate the appropriate template in Step 2.1 or Step 2.2 , then validate it here prior to NCBI submission: https://gmvs.fda.gov/
NCBI Pathogen package, customized for US labs doing enteric surveillance, including GenomeTrakr labs. This template has been widely used since 2013.
One Health Enteric Package: new metadata package available now for US labs doing enteric surveillance, including GenomeTrakr labs:
Custom, version-controlled template(s) available for download here: OHE GitHub page. OHE GitHub page.
- Our custom templates include extensive guidance and controlled vocabularies for most attributes.
- Sub-packages are available for download covering the major One Health samples types (human/animal hosts, food, food facilities, and farm/environment). Users can choose to populate the full package, or one more more of the sub-packages.
A generic version of this template was published by NCBI in 2022.
SRA sequence metadata template
Template for SRA metadata submission:
Download the generic "Metadata spreadsheet with sample names" file from the NCBI Submission Templates page:
https://submit.ncbi.nlm.nih.gov/templates/
And follow the guidance in the following table:
PRO TIPS:
- If you have sequences to submit that belong to more than one BioProject, create a separate submission + metadata table for each of your BioProjects.
- Entering fastq filenames in the spreadsheet : On a Mac, you can directly copy the file names from the folder into a spreadsheet. This is not possible on a PC using copy and paste but can be done with some command-line operation.
- Finally, it is important to develop a QA/QC step to make sure the files are associated with the correct sample name. For example, use a left function in excel to strip of the appended text in the file name and then use the exact match to make sure the name matches the sample name.
| A | B | C |
|---|---|---|
| Field | Description | Example |
| sample_name | Include the same ID here as you entered for "sample_name" in the BioSample submission template. Populate this field using the values in the PHA4GE specification for "specimen collector sample ID". | UT-12345 |
| library_ID | The library name should be a unique ID relevant to your workflow. It can be an autogenerated ID from your LIMS system or a modification of your sample_name. Populate this field using the values in the PHA4GE specification for "library_id". | UT-12345.6 |
| Title | Short, free text description that identifies the data on public pages. For Example: {methodology} of {organism}: {sample_name} | WGS of Salmonella enterica: UT-12345 |
| library_strategy | Overall sequencing strategy or approach. Choose from NCBI pick list | See NCBI SRA pick list. (e.g. WGS) |
| library_source | molecule type used to make the library | See NCBI SRA pick list. (e.g. Genomic) |
| library_selection | Library capture method | See NCBI SRA pick list. (e.g. random, PCR) |
| Library_layout | Choose from NCBI pick list | See NCBI SRA pick list, choose "paired" |
| platform | Sequencing platform | See NCBI SRA pick list. (e.g., Illumina). |
| instrument_model | Name of the sequencing instrument. | See NCBI SRA pick list. (e.g. Illumina MiSeq, iSeq 100) |
| Design_description | Free text description of methods | |
| Filetype | File format name for the raw sequence data Choose from NCBI pick list | See NCBI SRA pick list. (e.g. Fastq) |
| Filename | include ALL of the files resulting from this library. **Add additional fields if there are more than two files (e.g. Filename3). Populate this field using the values in the PHA4GE specification for "r1 fastq filename". | genome_r1.fastq (*must be exact) |
| Filename2 | genome_r2.fastq (*must be exact) Populate this field using the values in the PHA4GE specification for "r2 fastq filename". | genome_r2.fastq (*must be exact) |
| Filename3-8 | list other fastq file names (e.g. for NextSeq data) |
Save the second sheet (SRA_data) as a TSV (tab-delimited file) for upload in the “SRA metadata” tab within the submission portal.
*NCBI should also accept the original excel formatted file.

