Generation of full-length circRNA libraries for Oxford Nanopore long-read sequencing
Steffen Fuchs, Loélia Babin, Elissa Andraos, Chloé Bessiere, Semjon Willier, Johannes H. Schulte, Christine Gaspin, Fabienne Meggetto
Abstract
Circular RNA (circRNA) is a noncoding RNA class with broad implications for gene expression regulation, mostly by e.g. interaction with other RNAs or RNA-binding proteins. However, their specific sequence is not revealed by the commonly applied short-read Illumina sequencing. Here, we present an adapted protocol to enrich and sequence full-length circRNAs using the Oxford Nanopore long-read sequencing platform. The protocol involves an RNaseH-based ribodepletion, an enrichment of lowly abundant circRNAs by exonuclease treatment and negative selection of linear RNAs. Then, a cDNA library is created and amplified by PCR. This library is used as input for ligation-based sequencing together with native barcoding. Stringent quality control of the libraries is ensured by a combination of Qubit, Fragment Analyzer and qRT-PCR. The recommended amount of starting material is 7 µg of RNA, however lower amounts of RNA have also been tested successfully. Multiplexing of up to 4 libraries yields in total more than 1-2 Mio reads per library, of which 1-2 % are circRNA-specific reads with >99 % of them full-length. The protocol works well with human cancer cell lines. We further provide suggestions for Nanopore sequencing, the bioinformatic analysis of the created data, as well as the limitations of our approach and recommendations for troubleshooting and results interpretation. Taken together, this protocol enables reliable full-length analysis of circRNAs, a non-coding RNA type involved in a growing number of physiologic and pathologic conditions.
Steps
Enrichment of circRNAs for generation of Nanopore sequencing libraries
-
Ribodepletion
-
circRNA enrichment
-
cDNA library creation
-
Quality control
Suggestions for Nanopore sequencing and data analysis
-
Suggestions for Nanopore sequencing
-
Recommendations for bioinformatics analysis of the data
Expected results, limitations and troubleshooting
-
Expected results and interpretation
-
Limitations and challenges
-
Troubleshooting
The enrichment of the whole circRNA fraction follows the published workflow from Zhang et al . with several modifications:
-
Modification of the ribodepletion method from a commercial kit to the published method of Baldwin et al ., which is based on a pool of DNA oligonucleotides that hybridize with ribosomal RNA and a digest of DNA:RNA hybrids by RNaseH
CitationBaldwin A, Morris AR, Mukherjee N 2021 An Easy, Cost-Effective, and Scalable Method to Deplete Human Ribosomal RNA for RNA-seq. Current protocols https://doi.org/10.1002/cpz1.176 -
Clean-up steps and final size selection have been adapted to select for circRNAs longer than 200 nt and therefore also consider shorter circRNAs, whereas in the original protocol a selection of > 1kb was applied (the average length of circRNAs is between 200-800 nt)
-
Additional negative poly(A) selection for further enrichment of circRNAs
-
Increased quantity of retrieved library by using more starting material and a higher number of PCR cycles
-
Thorough quality control by combining qRT-PCR, Qubit and Fragment Analyzer after circRNA enrichment
Further, we refer to the used sequencing protocol for the Nanopore platform, suggest changes to the standard protocol and further give recommendations for the bioinformatics analysis. We pool 4 libraries. The enrichment workflow can therefore be performed in parallel for 4 samples.
Before start
Work in an RNase-free workspace. Clean your workspace and tools with a product, such as RNase Zap (Invitrogen, #AM9780).
Steps
1) Ribodepletion
Enrichment of circRNAs for generation of Nanopore sequencing libraries
This section provides the detailed protocol to enrich the fraction of circRNAs that will be used to create sequencing libraries for the Nanopore platform.
Ribodepletion
Hybridization and RNaseH treatment
Prepare the following: thaw 7µg total RNA On ice

Thaw one aliquot of rRNA depletion oligos On ice
Bring one aliquot of Agencourt RNA Clean XP beads to Room temperature at least 0h 30m 0s before use.
Set a heat block to 65°C
Set up the following PCR program in a thermocycler:
- 95°C - Hold (to heat up the thermocycler)
- 95°C - 3 min.
- Ramp: 0.1°C/s to 65°C
- 65°C - 5 min.
- 65°C - Hold (this is when the mixes will be put together)
- 65°C - 10 min.
Prepare the RNaseH mix and incubate it in the heat block ( step 3.3 ) at 65°C without adding the RNaseH enzyme :
| A | B | C |
|---|---|---|
| Reagent | Volume per Reaction [µl] | Final concentration |
| 5x RNaseH buffer | 4 | 1x |
| 100 mM MgCl2 | 6 | 10 mM |
| 5 U/µl RNaseH enyzme | 4 (add later) | 10 U |
| H2O | 6 | |
| Total volume | 20 |
Prepare the hybridization mix :
| A | B | C |
|---|---|---|
| Reagent | Volume per Reaction [µl] | Final concentration |
| 5x RNaseH buffer | 8 | 1x |
| 1 mM EDTA | 2 | 50 µM |
| rRNA depletion oligos (200 µM) | 9.9 | 30 µg |
| 7 µg of total RNA | x | 7 µg |
| H2O | fill up to 40 µl | |
| Total volume | 40 µl |
Incubate the sample for hybridization in the programmed thermocycler from step 3.4 until the 65°Chold is reached.
Add the RNaseH enzyme to the RNaseH mix from step 3.5 . Mix well by pipetting and spin down. Add 20µL of the RNaseH mix now with the RNaseH enzyme to the hybridization mix from step 3.6 . Mix well by pipetting and spin down. Place back in the thermocycler.
Continue the program of the thermocycler ( 65°C for 0h 10m 0s) then place the sample On ice
First bead clean-up
Prepare the following:
Leave RNA Clean XP beads for 0h 30m 0s at Room temperature before using them. Vortex thoroughly the RNA Clean XP beads until the solution is homogeneously brown.
Remove samples from the magnet, add 38.5µL H2O to the beads, resuspend well by pipetting. Incubate 0h 5m 0s at Room temperature .
Place the samples back on the magnet for 0h 5m 0s or at least until the beads are clearly separated from the supernatant.
Transfer 37.5µLof the supernatant to a new 0.2 ml tube (try not to transfer the beads).
Add 1x RNA Clean XP beads (60µL) to the RNase-H treated sample and mix well by pipetting until the solution is homogenous.
Incubate for 0h 5m 0sat Room temperature
Place sample on the magnet and incubate at Room temperature for at least 0h 5m 0sor until the beads are clearly separated from the supernatant. Discard supernatant without disturbing the beads.
Perform 2 washes with freshly prepared ethanol 80 % without disturbing the beads:
While leaving the sample on the magnet add 200µLEthanol 80 % without disturbing the beads.
Incubate for 0h 0m 30s at Room temperature. Remove the supernatant without disturbing the beads.
Repeat steps 3.6 and 3.7 .
Let samples air-dry (leave lid open) for 0h 5m 0s, take care not to over-dry the beads (overdrying is indicated by formation of cracks in the bead pellet).
Digestion of ribodepletion oligos by DNase
Prepare the DNase mix :
| A | B | C |
|---|---|---|
| Reagent | Volume per reaction [µl] | Final concentration |
| Turbo DNase (2U/µl) | 12 | 24 U |
| 10x DNase buffer | 5.5 | 1x |
| Total | 17.5 |
Add 17.5µLof DNase mix to the cleaned-up sample from before, mix well by pipetting and incubate at 37°Cfor 0h 30m 0s in a thermocycler.
Place 37On ice after incubation.
Second bead clean-up
Follow the procedure described for the first bead clean-up using 1x RNA Clean XP beads (55µL).
Elute in 15.5µL H2O.
Save 14.5µL of supernatant for circRNA enrichment. Put 37On ice.
2) circRNA enrichment
Polyadenylation of linear RNAs
Prepare the polyadenylation mix :
| A | B | C |
|---|---|---|
| Reagent | Volume per reaction [µl] | Final concentration |
| Ribodepeted RNA | 14.5 | |
| ATP (10 mM) | 2 | 1 mM |
| Poly(A) polymerase (5 U/µl) | 1 | 5 U |
| 10x reaction buffer | 2 | 1x |
| RNase-inhibitor (40 U/µl) | 0.5 | 20 U |
| Total volume | 20 |
Mix well by pipetting and spin down. Incubate 0h 30m 0s at 37°C in a thermocycler.
Clean-up with RNA Clean XP beads as described before, using a 1:1 ratio (20µLbeads).
Elute in 18µL H2O, save 17µL for RNaseR treatment. Put 37On ice
RNaseR treatment
Prepare the RNaseR mix :
| A | B | C |
|---|---|---|
| Reagent | Volume per Rx [µl] | Final concentration |
| Polyadenylated RNA | 17 | |
| 10x reaction buffer | 2 | 1x |
| RNaseR (20U/µl) | 0.5 | 10u |
| RNase inhibitor (40u/µl) | 0.5 | 20u |
| Total volume | 20 | **** |
Incubate for 0h 30m 0sat 37°C in a thermocycler.
Clean-up with RNA Clean XP beads as described before, using a 1:1 ratio (20µL beads). Elute in 21µL, save 20µL for negative poly-a selection. Put 37On ice.
Negative poly(A) selection
Bring the NEBNext® Poly(A) mRNA Magnetic Isolation Module toRoom temperature
Elute in 6µL, save 5µLfor cDNA library creation and quality control, keep 4On ice.
Vortex oligo(dT) beads until the solution is homogenous.
Take 20µL oligo(dT) beads in a separate PCR tube and wash twice with 100µLof RNA bead binding buffer by using a magnetic rack, resuspend the beads in 20µL of RNA bead binding buffer (1:1 ratio with RNA volume). Mix beads and RNA by pipetting.
Incubate 0h 5m 0s at 65°C and hold at 4°Cin a thermocycler.
Mix well by pipetting, leave 0h 5m 0s at 4Room temperature.
Mix well by pipetting, leave 0h 5m 0sat 4Room temperature.
Place tubes on the magnetic rack for 0h 5m 0s or until the liquid is clear.
Save the supernatant in a new PCR tube and keep 4On ice.
Clean-up with RNA Clean XP beads as described before, using a 1:1 ratio (40µL).
3) cDNA library creation
cDNA generation
Set up the following PCR program in a thermocycler:
- 3 min. at 72°C
- 10 min. at 25°C
- Hold at 42°C (this is when the reverse transcription mix gets added)
- 90 min. at 42°C
- 10 min. at 70°C
- Hold at 4°C
Prepare the hybridization mix :
| A | B | C |
|---|---|---|
| Reagent | Volume per Rx [µl] | Final concentration |
| RNA | 3.5 | |
| Custom SMARTer CDS Primer IIA (12 µM) | 1 | 2.7 |
| Total volume | 4.5 |
Mix well by pipetting. Incubate in the programmed thermocycler until the 42°C hold is reached.
In the meantime prepare the reverse transcription mix and add it to the hybridization mix , when the 42°C hold is reached:
| A | B | C |
|---|---|---|
| Reagent | Volume per Rx [µl] | Final concentration |
| 5x First-strand buffer | 2 | 0.36x |
| Dithiotreitol (DTT, 100 mM) | 0.25 | 4.5 mM |
| dNTP mix (10 mM) | 1 | 1.8 mM |
| SMARTer IIA oligo (12 µM) | 1 | 2.2 µM |
| RNase inhbitor (40 U/µl) | 0.25 | 10 U |
| SMARTScribe RT (100 U/µl) | 1 | 100 U |
| Total volume | 5.5 |
Mix well by pipetting, continue the PCR program until the 4°C hold is reached. Keep the sample On ice.
PCR amplification
Prepare the PCR mix :
| A | B | C |
|---|---|---|
| Reagent | Volume per Rx [µl] | Final concentration |
| SMARTer PCR primer (12 uM) | 6.8 | 0.8 |
| cDNA | 4 | |
| LongAmp Taq 2x Master mix | 50 | 1x |
| H20 | 39.2 | |
| Total | 100 |
Incubate using the following PCR program
| A | B | C |
|---|---|---|
| Step | Temperature [°C] | Time |
| Initial denaturation | 95 | 30 s |
| 25 cycles | 95 | 15 s |
| 62 | 15 s | |
| 65 | 2 min | |
| Final extension | 65 | 2 min |
| Hold | 4 |
Fragment size selection
Clean-up with AMPure XP beads (DNA-specific) as described before, using a 0.8:1 ratio (80µLbeads) to select for fragments > 200 nt. Elute in 31µLH2O, save 30µLfor creation of libraries for Oxford Nanopore sequencing and quality control. Keep 42On ice.
The cDNA library can be stored at -20°C for several weeks.
4) Quality control
Assessing the molarity of the library
Quantification of the library
Quantify the library by using a Qubit fluorometer with the BR dsDNA assay, following the manufacturer's instructions and using 1µL of amplified cDNA library.
Determination of the library size and quality
Check the average length of the library by e.g. Fragment Analyzer (hs NGS Fragment kit, 2µL of diluted amplified cDNA library as input) by following the manufacturer's instructions.
Calculation of the library's molarity
Calculate the amount of fmol in your sample by using the results of the quantification and library size determination. This can be done by using e.g. the Biomath Calculator from Promega (Link to Promega Biomath calculatores) using the calculator DNA: µg to pmol. The recommended input for Oxford Nanopore library preparation with the kit SQK-LSK109 is 1 µg (or 100-200 fmol) of PCR amplicons. Since the circRNA abundance is still relatively low, multiplexing of several libraries with the native barcoding kit (EXP-NBD104) is recommended to occupy enough pores.
Validation of circRNA enrichment
Use 1µL of enriched RNA from step 9.10 as input for reverse transcription using the Maxima H Minus RT kit (Thermo #K1682) without DNase treatment.
Prepare the reverse transcription mix :
| A | B | C |
|---|---|---|
| Reagent | Volume per Rx [µl] | Final concentration |
| Enriched RNA | 1 | |
| H2O | 12.5 | |
| Random hexamer primer (100 µM) | 0.5 | 2.5 µM |
| dNTP Mix (10 mM) | 1 | 0.5 mM |
| 5X RT Buffer | 4 | 1x |
| Maxima H Minus enzyme mix | 1 | |
| Total volume | 20 |
Mix by pipetting. Program a thermocycler and incubate the sample as follows:
- 10 min at 25 °C
- 15 min at 50 °C
- 5min at 85°C
- Hold at 4°C
Dilute 1µLof cDNA 1:10 and use it as input for qRT-PCR using the FastStart Essential DNA Green Master 2x (Roche #06402712001).
Prepare the qRT-PCR mix for each target RNA:
| A | B | C |
|---|---|---|
| Reagent | Volume per Rx [µl] | Final concentration |
| FastStart Essential DNA Green Master 2x | 5 | 1x |
| Forward primer [10 µM] | 0.5 | 0.5 µM |
| Reverse primer [10 µM] | 0.5 | 0.5 µM |
| H20 | 3 | |
| cDNA | 1 | |
| Total volume | 10 |
Run for 40 cycles with the following PCR program:
| A | B | C |
|---|---|---|
| Step | Temperature [°C] | Time |
| 50 | 2 min | |
| 95 | 10 min | |
| 40 cycles | 95 | 15 s |
| 60 | 60 s | |
| Run melt curve analysis |
5) Suggestions for Nanopore sequencing
Suggestions for Nanopore sequencing and data analysis
This section provides links to the used Nanopore sequencing protocols with further information and improvements, and suggestions for the bioinformatics analysis.
Protocols for Oxford Nanopore sequencing:
Protocols are available from the Nanopore community (free login required):
The following protocol was used according to the manufacturer:
Native barcoding amplicons (with EXP-NBD104, EXP-NBD114, and SQK-LSK109) v12Nov2019
We included small modifications that we will describe below together with the general steps of the library preparation. We use an Oxford Nanopore MinION MK1C sequencer that includes a graphical card needed for base calling of the raw Nanopore sequencing data (.fast5 format) to generate .fastq files that are needed for further analysis. A standard MinION can be used as well and base calling can be performed afterwards with a computer and the Nanopore MinKNOW software.
1) End-preparation:
This step prepares the DNA ends for adapter attachment. The manufacturer recommends 100-200 fmol of RNA input. We use up to 1000 fmol, to have enough material for several rounds of sequencing. Our circRNA-enrichment workflow provides enough output to achieve this.
2) Native barcode ligation and pooling:
Nanopore barcodes will be attached to the ends of the DNA in this step. The manufacturer recommends to use 100-200 fmol of end-prepped DNA. Here, we used 22.5 µl of the reaction from step 1). The samples are cleaned-up with magnetic beads and eluted. The manufacturer recommends a high elution volume, which would make a concentration step necessary whenmultiplexing various samples. To avoid this, we elute only in 11 µl, to obtain 10 µl of sample for the pooling and 1 µl to measure the concentration (see below).
For the pooling, we measure the concentration of the sampleswith a Qubit fluorometer (BR dsDNA assay) and calculate the molarity as described above. Expected recovery aim 15-25 ng/µl (molarity 30-70 fmol/µl). Equimolar amounts of samples should be pooled. The protocol recommends 100-200 fmol, we usually use 400 fmol per sample and pool 4-5 samples.
3) Adapter ligation and clean-up
This step attaches the Nanopore sequencing adapters. After adding the adapters a clean-up is performed with magnetic beads. The manufacturer uses a ratio of 0.5x beads to sample to select for long fragments >500 nt. However, we use a ratio of 0.8x to include as well fragments of >200nt, since the expected size of circRNAs is 200-800 nt (Guo, 2014; Zheng, 2019). This also is the reason why the short fragment buffer has to be used for the clean-up. We perform in total 3 washes with the short fragment buffer to eliminate chemical components that might potentially interfere with sequencing (only 2 are recommended in the official protocol). The cleaned-up sequencing pool is measured with a Qubit fluorometer (BR dsDNA assay). Recovery aim: 30-50 ng/µl (ca. 50-90 fmol/µl). To calculate the molarity, we use the average length (as measured in the QC section by Fragment Analyzer) of all samples pooled. The final library pool is ready for sequencing and should be stored for short-term at 4 °C until the sequencing run is finished for possible flushing of the flow cell and reloading of the library (see below).
4) Priming and loading the flow cell
Finally the flow cell is primed and the library loaded. Please note that the pores in the flow cell needed for sequencing are not very stable and the number of active pores correlates with the sequencing output. Care has to be taken to keep as much pores active for sequencing as possible. For instance, air bubbles that get introduced by pipetting during the priming process can irreversibly damage them. Further, during priming the storage buffer that keeps the pores stable is taken off. The manufacturer recommends to wait 5 min. until the library gets loaded. In our experience it is best to prepare the library mix before starting the priming process so that the pores are as short as possible without the storage buffer.
The recommendation of the manufacturer is to load 5-50 fmol. In our experience this can be increased to 200 fmol. Of note, this does still not fully saturate the pores.
Sequencing is started on the MK1C with the base calling option activated using standard settings. We further activate also demultiplexing (standard settings), to directly separate the reads in the respective samples.
6) Recommendations for bioinformatics analysis of the data
Bioinformatics analysis
Demultiplexing and base calling of raw data were realized by using the Guppy toolkit from the MinKNOW software (v22.05.8) using standard parameters as described above. This creates .fastq files from the raw .fast5 data. Only passed .fastq files assigned to samples were used for next analyses.
Analysis of the base-called sequencing data in fastq format involves the following steps:
• Cleaning the reads from the adapters with cutadapt v3.4
• Identifying circRNAs by using the CIRI-Long tool v1.0.3
• Creating alignments with minimap2 v2.19 and visualize circRNA-spanning reads in IGV Genomics Viewer v2.9.4.
The used genome annotation was GRCh38.
Cleaning the reads
Cleaning the reads was done with cutadapt v3.4 and removes the adapter sequences. The following settings were used for the analysis:
-
Select reads with non-zero length (-m 1) and set less than 20 % error rate (-e 0.2)
-
Command lines to clean the reads with the used adapter sequences:
cutadapt -e 0.2 -m 1 -g AAGCAGTGGTATCAACGCAGAGTAC -o FileName_5pTrimmed.fastq.gz FileName.fastq.gz
cutadapt -e 0.2 -m 1 -a GTACTCTGCGTTGATACCACTGCTT -o TrimmedFileName.fastq.gz FileName_5pTrimmed.fastq.gz
Identifying circRNAs
We used the CIRI-long software (v1.0.3; Zhang, 2021) with default parameters to identify circRNAs. CIRI-long first splits reads produced by rolling circle amplification into repetitive fragments by searching for identical repeated sequences. This first step allows to detect the boundaries of circRNAs which are then aligned against the genome to generate a consensus sequence. It uses splice sites from known exon annotations and canonical de novo GT/AG splice signals to align junction sites but also non-canonical splice signals when canonical signals are not present. Of note, a bwa index is required for the reference genome.
Only passed reads, not considered unclassified, were used for the analysis.
- Command lines used:
CIRI-long call -i FileName.fastq.gz -o step1 -r genome.fa -p SampleName -a genome.gtf -t 10
CIRI-long collapse -i SampleFile.lst -o step2 -p SampleName -r genome.fa -a genome.gtf -t 10
CIRI-long will create several output files with the distinct circRNA isoforms detected and the number of reads in the different samples.
In step1, it provides fasta files of reads predicted as circRNA with related information on, for example, the size and genomic coordinates of the circRNA, and the identified splice signal. Isoforms from all samples are collapsed in step2. The main outputs are an expression matrix providing collapsed isoforms counting and a gtf-like file that contains detailed information of circRNAs and annotation of circRNA back-spliced regions.
Alignments to visualize circRNA-spanning reads
CIRI-Long involves an alignment step, but the bam files containing the aligned reads are not conserved. Therefore, we created separate alignments, by aligning long reads against the human genome (GRCh38) using the minimap2 software (v2.19, Li, 2018), converted in bam format by using samtools (v1.12, Li, 2009) and visualized using IGV Genomics Viewer (v2.9.4, Robinson, 2011) keeping supplementary alignments, which allows to visualize inverted segments.
- Command lines used:
minimap2 -ax splice -t 20 -uf -k14 genome_index FileName.fastq.gz > FileName.sam ; samtools view -b FileName.sam|samtools sort > FileName.bam ; samtools index FileName.bam).
7) Expected results and interpretation
Expected Results, limitations and troubleshooting
Expected results and interpretation
Using our protocol libraries of an average length of 606.75 nt and a concentration of 5 ng/µl, thus almost 150 ng in total can be generated (Fig. 2, Tab. 2). This will be enough for several rounds of sequencing, if a high number of reads is needed to also detect lower expressed circRNAs. The library length is about the average published size of circRNAs, which is reported to be between 200 - 800 nt (Guo, 2014; Zheng, 2019) and shows that our workflow does not fragment RNA and maintains its size.
| A | B | C | D | E | F |
|---|---|---|---|---|---|
| **** | SU-DHL-1 | Karpas-299 | COST | SUP-M2 | Average |
| Concentration [ng/µl] | 5.12 | 4.68 | 4.92 | 4.5 | 4.81 |
| Size [nt] | 654 | 629 | 573 | 571 | 606.8 |
Table 2: Results of the library preparation. Libraries for Nanopore sequencing were prepared of 4 different anaplastic large-cell lymphoma cell lines (SU-DHL1, Karpas-299, COST, SUP-M2).The concentration was measured with the Qubit BR dsDNA kit and library size by Fragment Analyzer with the hs NGS kit.
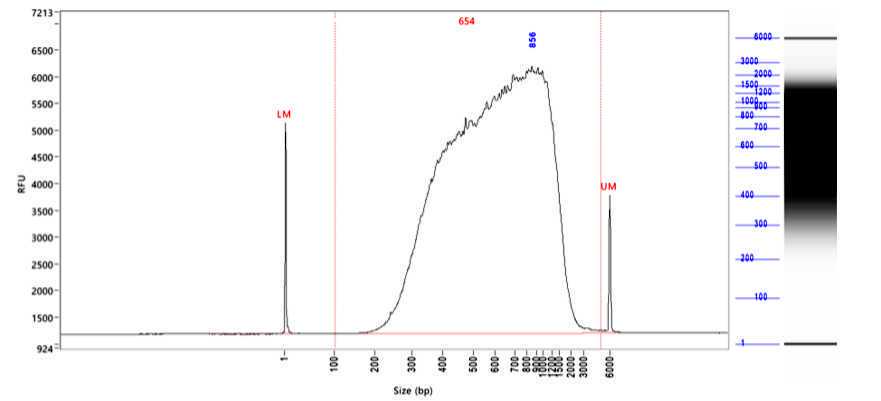
Figure 2: Generated libraries have the size of the average circRNA length. Shown is the library created from RNA of the anaplastic large-cell lymphoma cell lineSU-DHL-1. The library size was analyzed by Fragment Analyzer with the kit hs NGS. The average library size was 654 nt. RFU, relative fluorescence units.
Unwanted RNA transcripts should get depleted and the depletion is tested by qRT-PCR. Ribosomal RNA, as indicated by 18S rRNA, gets usually depleted more than 5 Cts, we achieve regularly a depletion of around 10 Ct. Further, mitochondrial RNA (as indicated by mtRNR1), small-nucleolar RNA (as indicated by RNU6B) and the RNA component of the signal recognition particle (as indicated by RN7SL2), which can be very abundant, should be depleted > 5 cycles. circRNAs should be stable or enriched, and cognate linear RNAs should get depleted (Fig. 3).
 were treated enzymatically to enrich for circRNAs as described in the protocol. The expression of circRNAs and unwanted transcripts (ribosomal RNA, 18S rRNA; mitochondrial RNA, mtRNR1; small-nucleolar RNA, RNU6B; signal recognition particle RNA, RN7SL2 and linear RNAs/mRNAs, linkZKSCAN1, linHIPK3) was analyzed by qRT-PCR and compared with an untreated Mock control.")
Using the ligation-based sequencing kit (SQK-LSK109) together with the native barcoding kit (EXP-NBD104) 4 libraries of 4 different human anaplastic large-cell lymphoma cell lines (SU-DHL-1, Karpas-299, COST, SUP-M2) were pooled together and the pool was sequenced on a MinION (MK1C, Oxford Nanopore). The sequencing output was on average 1,536,242 reads per library and reads were of high quality (Tab. 3, mean Q-score 15).
| A | B | C | D | E | F |
|---|---|---|---|---|---|
| **** | SU-DHL1 | Karpas-299 | COST | SUP-M2 | Average |
| Raw reads | 1,473,419 | 1,734,196 | 899,725 | 2,037,577 | 1,536,229 |
| Mean read length [nt] | 459.6 | 368.2 | 386.3 | 403.3 | 404.4 |
| Maximum read length [nt] | 4,006 | 3,889 | 3,538 | 3,455 | 3,722 |
| BSJ-reads [% of reads] | 1.05 | 0.95 | 0.95 | 1.06 | 1.00 |
| Full-length circRNAs | 15,673 | 16,725 | 8,750 | 21,918 | 15,767 |
| Different circRNAs | 3,143 | 3,195 | 1,426 | 4,017 | 2,945 |
| Mean circRNA length [nt] | 435.1 | 354.9 | 366.4 | 370.4 | 381.7 |
| Maximum circRNA length [nt] | 1,798 | 1,634 | 1,596 | 2,228 | 1,814 |
Table 3: Sequencing results obtained with one MinION flow cell. circRNA-enriched libraries from 4 anaplastic large-cell lymphoma cell lines were sequenced by Oxford Nanopore. Calculations are based on the passed reads and the circRNA analysis was performed with CIRI-Long.
The run took on average 40 h. Of note, the pores were not completely saturated, so probably a longer sequencing run with higher output would have been possible. Following the analysis workflow described for CIRI-Long we could identify on average 15,767 circRNA-specific reads, thus 1.0 % of the total reads, similar to the study from Zhang et al. , of which 99 % covered the full length of the circRNA. For most of them, concatamers were detected and, as expected, several isoforms were identified and reported by CIRI-Long. On average 2,945 different circRNAs were identified. We noticed that the more reads are generated, more different full-length circRNA isoforms are detected, which could be another argument for deeper sequencing. The results were comparable among the samples from the 4 different cell lines, showing the robustness of the workflow.
In summary, this modified protocol facilitates consistent full-length sequencing of circRNAs, which will help to study this noncoding RNA type in a variety of physiologic and pathologic contexts.
8) Limitations and challenges
Limitations and challenges
A limitation of the protocol is the relatively high input of RNA of 7 µg. While this amount of RNA worked best in our experiments, we also tried successfully only 3-5 µg. Further, in general the Nanopore sequencing platform produces less reads than Illumina-based short read techniques, especially when using the MinION. That means lowly abundant circRNAs might not be detected by our sequencing protocol.
Limitations of CIRI-Long are that the alignment parameters cannot be modified and bam files containing the aligned reads are not conserved. Further, there is no option to detect fusion circRNAs derived from fusion genes (distant genes, genes located on different chromosomes).
9) Troubleshooting
Troubleshooting
Below we provide assistance and recommendations for problems that can occur during this protocol and the analysis of the data.
circRNA enrichment
- Step 7 RNaseR treatment: if the enrichment of circRNAs seems to be insufficient, a longer incubation with RNaseR could be tried.
- Step 11 PCR amplification: if the obtained amount of cDNA is much lower than expected, the volume of the PCR reaction and/or the amount of PCR cycles could be increased.
- Step 12 Fragment size selection: if the size of the circRNAs of interest that should be enriched is much higher or lower, the ratio of beads to DNA could be adapted. A higher ratio will lead to retention of smaller fragments, a lower ratio will select for longer fragments.
Validation of circRNA enrichment
- While we provide a selection of primers covering different RNA species that should be depleted by the workflow, further transcripts can be checked by qRT-PCR in this step.
Nanopore sequencing
- Care has to be taken to not introduce air bubbles while priming or loading a flow cell, since this will damage pores irreversibly.
- The pore activity should be closely monitored while sequencing. If a lot of inactive pores accumulate, a flow cell wash could be performed as described above.
- If a lot of pores are available, but not occupied, more library could be loaded. If the translocation speed is > 300, then no further priming is usually needed.
Bioinformatics analysis
- Alignments were re-generated using minimap2 (v2.19, GRCh38) and visualized in IGV Genomics Viewer (v2.9.4) to validate the presence of concatamers. Linking supplementary alignments also helped to visualize circularization junctions.
- To detect fusion circRNAs sam files produced by minimap2 during the alignment can be filtered to conserve chimeric alignments that contain segments of the same read aligning to distant genes (on the same or different chromosomes).
