Further Micro-scaled MEDI (Macronutrient Extraction and Determination from Invertebrates)
Jordan P Cuff
macronutrient
nutritional
nutrient
invertebrate
insect
colorimetric assay
entomology
entomological
lipid
protein
carbohydrate
Disclaimer
DISCLAIMER – FOR INFORMATIONAL PURPOSES ONLY; USE AT YOUR OWN RISK
The protocol content here is for informational purposes only and does not constitute legal, medical, clinical, or safety advice, or otherwise; content added to protocols.io is not peer reviewed and may not have undergone a formal approval of any kind. Information presented in this protocol should not substitute for independent professional judgment, advice, diagnosis, or treatment. Any action you take or refrain from taking using or relying upon the information presented here is strictly at your own risk. You agree that neither the Company nor any of the authors, contributors, administrators, or anyone else associated with protocols.io, can be held responsible for your use of the information contained in or linked to this protocol or any of our Sites/Apps and Services.
Abstract
Macronutrients, comprising carbohydrates, proteins and lipids, underpin many ecological processes, but their quantification in ecological studies is often inaccurate and laborious, requiring large investments of time and bulk samples, which make individual‐level studies impossible. This is a protocol for the direct, rapid and relatively low‐cost determination of macronutrient content from single small macroinvertebrates.
Macronutrients are extracted by a sequential process of soaking in 1:12 chloroform:methanol solution to remove lipid and then solubilising tissue in 0.1 M NaOH. Proteins, carbohydrates and lipids were determined by colorimetric assays from the same individual specimens.
Macronutrient Extraction and Determination from Invertebrates (MEDI) can directly and rapidly determine macronutrient content in tiny (dry mass <1 mg). Using MEDI, the total macronutrient content of over 50 macroinvertebrates can be determined within around 3 days of collection at a cost of ~$1.35 per sample.
Before start
Please read the full protocol before starting and ensure that you have all the necessary equipment/reagents. Particular attention must be paid to the COSHH and safety documentation relating to the reagents. The original MEDI protocol is recommended for samples with dry masses greater than 1 mg. For samples which are borderline, consider using the original protocol before this adaptation; it will likely produce more accurate results.
Steps
Welcome to MEDI!
Welcome to Macronutrient Extraction and Determination from Invertebrates (further micro-scaled edition)!
The following presents a micro-scaled version of the original MEDI protocol. The further micro-scaled protocol is intended particularly for invertebrates of dry volumes of 1 mg or less which cannot be pooled together given their scarcity or the focus of the study precluding pooling (e.g., investigation of individual differences). The micro-scaling is achieved by increasing analyte concentration at both the extraction and analysis stages, therefore facilitating more sensitive detection of macronutrients. Analytical standards must also be adjusted accordingly (detailed within).
Here's an overview of the further micro-scaled extraction protocol:

This is based on the protocol originally published in Methods in Ecology and Evolution:
There are several overnight incubation steps, so don't be alarmed by some of the long procedure times!
Collection and preparation of materials
Collect invertebrates and kill them, ideally by freezing or another optimally humane method.
Desiccate the invertebrate specimens via freeze-drying (ideal) or heat-drying until completely dry.
If possible, weigh the dried invertebrate using appropriately sensitive scales, recording the mass.
Determination of lipid content
Add 500 μl of 1:12 chloroform:methanol to each sample tube and leave the tube containing the specimen and chloroform:methanol at room temperature for 24 hours.
Remove 400 μl of chloroform:methanol solution and keep it in a separate tube for lipid analysis. The remaining solution in the original tube is taken forward to the next section (determination of protein and carbohydrate content; STEP 15), ideally with immediacy given that the next step is another overnight soak in chloroform:methanol.
The lipid content of the 400 μl of chloroform:methanol solution removed during STEP 6 can be determined using the sulfo-phospho-vanillin method.
Prepare a stock standard dilution series using a suitable analogue to the lipids present in your tissues (e.g. analytical lard oil for animals) of known concentration diluted with 1:12 chloroform:methanol. A dilution series of 0-1 mg/ml in nine increments (0, 12.5, 62.5, 125, 250, 375, 500, 750 and 1000 μg/ml) should suitably cover a broad range of concentrations, and can be adjusted further according to expected concentrations.
Make up the vanillin reagent using the following amounts (which should account for reagent overage) per sample/standard repeat (so thrice per sample/standard if running triplicates, as advised):
| A | B |
|---|---|
| Reagent | Amount |
| Vanillin | 275 μg |
| Hot water | 46 μl |
| 85 % phosphoric acid | 183 μl |
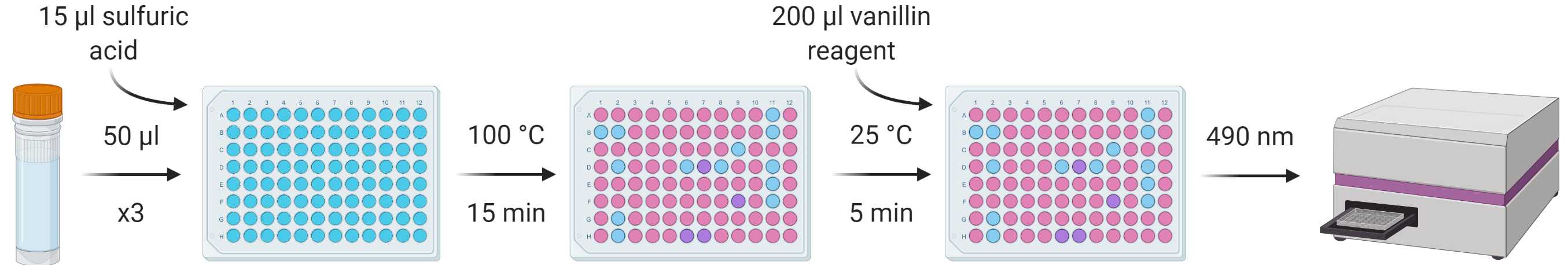
From each standard and sample, put three repeats of 50 µl into a flat-bottomed 96-well plate and heat at 100 °C in a ventilated hood until all solvent has evaporated (leaving just the lipid residue; ~5-10 min).
Add 15 µl concentrated sulfuric acid to the lipid residues and vortex/mix before incubating again at 100 °C for 15 min.
Allow the samples to cool to room temperature and add 200 µl vanillin reagent to each well, vortexing/mixing for homogenous colouration.
After 5 min, transfer 200 µl from each well into a corresponding well in a new plate.
Measure absorbance at 490 nm using a spectrophotometer.
Determination of protein and carbohydrate content
This continues with material generated in STEP 6.
From the original tube containing the remaining chloroform:methanol solution and the invertebrate tissue, discard the remaining ~100 μl supernatant (having already removed 400 μl for lipid analysis) avoiding the removal of any tissue. Add another 500 μl of 1:12 chloroform:methanol to each sample tube and leave the tube containing the specimen and chloroform:methanol at room temperature for 24 hours.
Discard the supernatant, avoiding the removal of any tissue, and evaporate any remaining residual solvent.
Lyse the tissue and ensure it is mixed/homogenous. Lysis can be carried out using a pestle and mortar, or bead-beating method, ensuring all available tissue is taken forward and any beads added are removed.
Add 500 μl 0.1 M NaOH to the sample tube. Incubate at 80°C for 30 min in a themo-shaker to ensure mixture of the lysed material with the NaOH.
Allow the sample to soak overnight at room temperature.
Centrifuge the sample for 10 min at 13,000 rpm.
Transfer 400 µl of the supernatant into a separate tube for taking forward to protein and carbohydrate determination.
The carbohydrate content of the 400 μl of NaOH solution removed during STEP 34 can be determined using the anthrone method.
Prepare a stock standard dilution series using a suitable analogue (e.g. analytical corn starch) of known concentration diluted with polished water. A dilution series of 0-0.02 mg/ml in nine increments (0, 0.25, 1.25, 2.5, 5, 7.5, 10, 15 and 20 μg/ml) should suitably cover a range of minute concentrations, but this can be adjusted accordingly. These concentrations were used for analyses of relatively carbohydrate-poor taxa like parasitoid wasps, thus more concentrated standards, e.g., a factor of ten more, may be better for other studies. Carbohydrate is typically much less concentrated in many invertebrate tissues compared to protein and lipid.
Make up the anthrone reagent using the following amounts (which should account for reagent overage) per sample/standard repeat (so thrice per sample/standard if running triplicates, as advised):
| A | B |
|---|---|
| Reagent | Amount |
| Anthrone | 165 mg |
| Concentrated sulfuric acid | 165 μl |
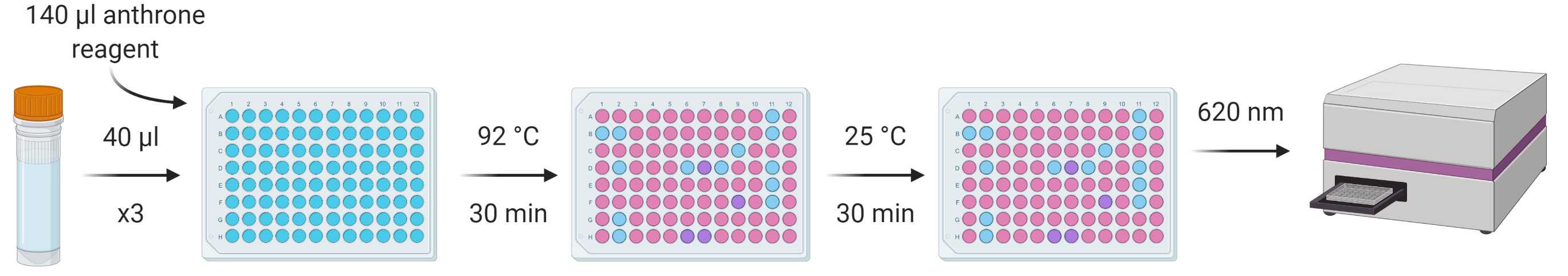
From each standard and sample, put three repeats of 40 µl into a flat-bottomed 96-well plate, add 140 µl anthrone reagent to each and vortex/mix.
Incubate the plate at 92 °C in a ventilated hood for 30 min.
Cool the plate to room temperature and incubate for a further 30 min to facilitate further development of the assay.
Measure absorbance at 620 nm using a spectrophotometer.
The protein content of the 400 μl of NaOH solution removed earlier can be determined using one of several colorimetric protein methods. The three highlighted in the MEDI manuscript (the benefits and drawbacks also discussed therein) are all viable, as are several other alternatives. This protocol will describe the protocol for the Lowry assay.
Prepare a stock standard dilution series using a suitable analogue (e.g. bovine serum albumin) of known concentration diluted with polished water. A dilution series of 0-1 mg/ml in nine increments (0, 12.5, 62.5, 125, 250, 375, 500, 750 and 1000 μg/ml) should suitably cover a broad range of concentrations, but this can be adjusted accordingly.
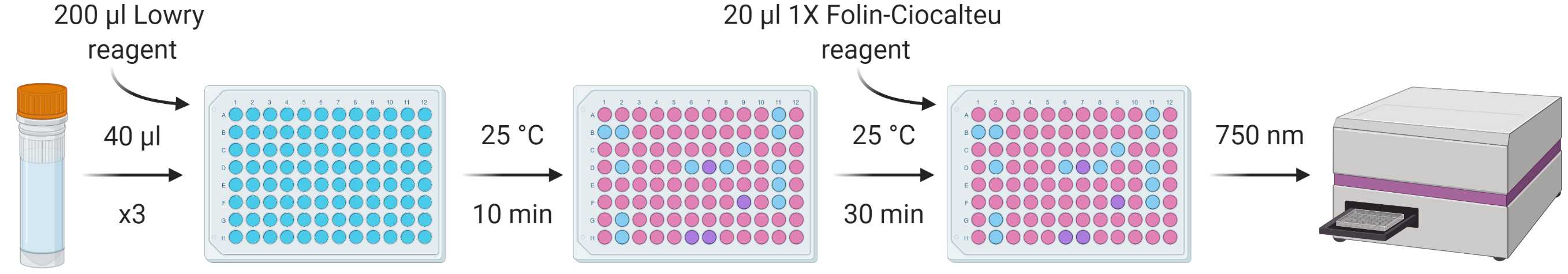
From each standard and sample, put three repeats of 40 µl into a flat-bottomed 96-well plate, add 200 µl of Modified Lowry Reagent to each and vortex/mix.
Incubate at room temperature (~20-25 °C) for 10 min.
Add 20 µl of 1X (1N) Folin-Ciocalteu reagent to each well and vortex/mix.
Incubate at room temperature (~20-25 °C) for 30 min.
Measure absorbance at 750 nm using a spectrophotometer.
Analysis of absorbance readings
You can now calculate the concentration of analyte based on the absorbance measurements. Keep in mind that the use of only 500 µl in each of the extraction phases means that mg/ml data will need dividing by two in order to calculate whole body macronutrient content. Any dilutions or other alterations to assay input concentrations/volumes must also be considered here.
