Bulk FLASH-seq
Simone Picelli, Vincent Hahaut, Rebecca A Siwicki
Abstract
Bulk RNA sequencing has revolutionized the study of transcriptomes, enabling the analysis of gene expression in complex tissues and heterogenous cell populations. While single cell RNA sequencing (scRNA-seq) has gained popularity due to its ability to profile individual cells, it comes with limitations such as high costs and reduced sensitivity for detecting low-abundance transcripts.
Here, we present bulk FLASH-seq (FS), a full-length RNA-seq method based on the single cell FLASH-seq workflow (Hahaut et al , 2022, https://www.nature.com/articles/s41587-022-01312-3), updated for bulk RNA analysis. FS bulk generates high quality data while requiring minimal hands-on time and offering a greater degree of customization. As a homebrew protocol, it is inexpensive compared to commercial kits allowing you to invest in greater sequencing depths or in a higher number of sequenced samples.
Our protocol enables comprehensive transcriptome analysis of bulk RNA samples, providing an alternative approach to scRNA-seq for gene expression when single-cell RNA-sequencing is not required.
Before start
The protocol should be carried out in a clean environment, ideally on a dedicated PRE-PCR workstation or on a separate bench used only for this purpose. Before starting, clean the bench and wipe any piece of equipment with RNAseZAP or 0.5% sodium hypochlorite. Rinse with nuclease-free water to avoid corrosion of delicate equipment.
Work quickly and preferably on ice.
Reagent mixes should be prepared shortly before use.
Mix thoroughly each mix before dispensing. For higher accuracy use liquid handling robots and/or nanodispensers whenever possible. For FLASH-Seq we use the I.DOT (Dispendix) for all the dispensing steps and the Fluent 780 liquid handling robot (Tecan) for sample cleanup, reagent transfers and pooling.
Always use LoBind plates and tubes (especially for long-term storage) to prevent the cDNA/DNA from sticking to plastic.
Steps
Prepare Lysis Mix
Prepare the following Lysis Mix:
| A | B | C | D |
|---|---|---|---|
| Reagent | Reaction concentration | Volume (µl) | Volume 384-well plate (μl) |
| Triton-X100 (10% v/v) | 0.2% | 0.020 | 9.216 |
| dNTP mix (25 mM each) | 6 mM | 0.240 | 110.592 |
| SMART dT30VN (100 µM) | 1.8 µM | 0.018 | 8.294 |
| RNase Inhibitor (40 U/μl) | 1.2 U/µl | 0.030 | 13.824 |
| DTT (100 mM) | 1.2 mM | 0.012 | 5.530 |
| dCTP | 9 mM | 0.090 | 41.472 |
| Betaine | 1 M | 0.200 | 92.160 |
| Nuclease Free Water | NA | 0.068 | 31.334 |
| Total volume | 0.678 | 312.422 |
Add 0.68µL lysis mix to each well of a 384-well plate.
Seal the plate with a PCR seal and quickly spin down to collect lysis mix at the bottom of the wells.
Process immediately to the next step or store plate long term at -20°C. Plates that will be used the same day can be stored in the fridge 4°C or on ice.
Safe stopping point . Plates containing lysis buffer can be stored for 6+ months at -20°C
Sample Normalisation & Addition
Quantify input RNA using a spectrophotometry and fluorometry assay (ex: NanoDrop™ or Qubit™ RNA High Sensitivity).
Normalise input RNA to 2 ng/μL with Nuclease-free water.
Add0.5µL of normalised RNA into corresponding 384-well plate containing lysis mix.
Seal the plate with an aluminium seal, quickly spin down. If processing more than one plate at once, keep each plate on dry ice until ready to transfer all to -80°Cfor long term storage. Plates containing RNA should ideally be processed within 6 months.
Cell Lysis
Remove plates from -80°Cand check that aluminum seal is still intact. If damaged or not sticking to the plate, wait a few minutes for the plate to partially thaw, remove damaged foil and replace with new one.
Place plate in a thermocycler with a heated lid and incubate for 0h 3m 0s minutes at72°C, followed by a 4°C hold step.
Spin down any condensation droplets (0h 0m 30s, 750x g ) that may have formed during incubation and return the plate to a cold block. Process quickly to the next step. If not ready with RT-PCR mix, keep the plate on the cold block at all times.
RT-PCR Reaction
Prepare RT-PCR mix:
| A | B | C |
|---|---|---|
| Reagent | Volume (μl) | Volume 384-well plate (μl) |
| DTT | 0.238 | 109.670 |
| MgCl2 (1M) | 0.046 | 21.197 |
| Betaine (5M) | 0.800 | 368.640 |
| Takara RNAse inhibitor (40 U/µl) | 0.096 | 44.237 |
| Maxima RT or Superscript IV (200U/µl) | 0.050 | 23.040 |
| KAPA HiFi HotStart Ready Mix (2X) | 2.500 | 1152.000 |
| FS TSO | 0.092 | 42.394 |
| Total volume | 3.822 | 1792.512 |
Add 3.822µL RT-PCR mix into each well of the 384-well plate.
Seal the plate with a PCR seal, gently vortex and spin down (0h 0m 30s, 750x g) to collect the liquid at the bottom of the well.
Place plate in a thermocycler with a heated lid and start the following RT-PCR program:
| A | B | C | D | E |
|---|---|---|---|---|
| Step | Temperature | Time | Cycles | |
| RT | 50°C | 60 min | 1 | |
| PCR | initial denaturation | 98°C | 3 min | 1 |
| denaturation | 98°C | 20 sec | 12-14x | |
| annealing | 67°C | 20 sec | ||
| elongation | 72°C | 6 min | ||
| 4°C | hold |
*Adjust the number of cycles according to the amount of RNA used.
Safe Stopping Point . Amplified cDNA before purification can be stored for several months at -20°C .
Magnetic bead working solution preparation
You can either use AMPure XP beads, SPRI beads or prepare your own solution of SeraMag beads containing 18% w/v PEG to reduce costs. A detailed protocol for making your own magnetic bead solution is described in:
| A | B | C |
|---|---|---|
| Reagent | Final concentration | Amount to add |
| Sodium chloride | 1 M | 2.92 gr |
| Tris-HCl, pH = 8.0 (1 M) | 10 mM | 500 µl |
| EDTA (500 mM) | 1 mM | 100 µl |
| PEG (MW=8000) | 18% w/v | 9.5 gr |
| Nuclease-free water | - | to a final volume of 50 ml |
Combine the sodium chloride, Tris-HCl, EDTA and PEG in a a 50-ml Falcon tube. Add 25mL water.
Solubilise the PEG by stirring and heating the solution at 37°C. If needed, progressively add more water.
While the PEG is dissolving, prepare the Sera-Mag SpeedBeads™. Vortex thoroughly to ensure complete resuspension. Withdraw1mL Sera-Mag SpeedBeads™ stock solution and transfer it into a new 1.5-ml tube.
Pellet the beads by placing the tube on a magnetic stand. Wait until the solution clears and then discard the supernatant.
Add 1mL TE buffer (10 mM Tris–HCl pH 8.0 + 1 mM EDTA) and resuspend the beads off the magnet.
Pellet the beads again, wait until the solution is clear, discard the supernatant and resuspend off the magnet with 0.9mL TE buffer.
Once the PEG solution is clear, add the resuspended beads prepared in the previous step.
Add 50µL Tween-20 (10% v/v) and 250µL sodium azide (NaN3, 10% w/v) and adjust the volume to 50 ml with nuclease-free water.
Store at 4°C. Do not freeze.
cDNA purification
Remove the Sera-Mag SpeedBeads™ working solution (AMPure XP beads or SPRI beads when using a commercial solution) from the 4°C storage and equilibrate it at room temperature for 0h 15m 0s.
Add 0.8X volume ratio Sera-Mag SpeedBeads™ working solution to each well. Mix thoroughly by pipetting or vortexing.
Incubate the plate off the magnetic stand for 0h 5m 0s at Room temperature.
Place the plate on the magnetic stand and leave it for 0h 5m 0s or until the solution appears clear.
Remove the supernatant without disturbing the beads. Do not let the bead pellet dry completely as it can decrease the final cDNA yield.
Remove the plate from the magnetic stand, add 15µL nuclease free water and mix well by pipetting or vortexing to resuspend the beads.
Incubate for 0h 2m 0s off the magnetic stand.
Place the plate back on the magnetic stand and incubate for0h 2m 0sor until the solution appears clear.
Remove 14µL of the supernatant and transfer it to a new plate.
Safe stopping point . Amplified and clean cDNA can be stored for several months at -20 C. We recommend storing in LoBind plates to avoid material loss during long-term storage.
Quality control check (highly recommended)
Check the cDNA quality on the Agilent Bioanalyzer High Sensitivity DNA chip. A good sample is characterised by a low proportion of fragments <400 bp, absence of residual primers (~100bp) and an average cDNA size of 1.8-2.2 Kb.
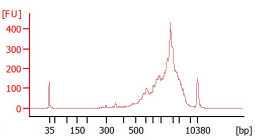
cDNA quantification (optional but recommended)
Allow the Quant-iT™ PicoGreen™ dsDNA Assay reagents to come to room temperature before opening the vial. PicoGreen™ dye is light sensitive, it should be thawed in dark drawer or wrapped in foil.
This step can be performed in either 384-well or 96-well plates. Volume of PicoGreen + TE solution should be adjusted depending on plate type.
Prepare a 1X TE solution using TE 20X (supplied) and nuclease-free water.
Prepare the standard curve using Lambda DNA standard (supplied at concentration of 100 ng/μl, with the PicoGreen™ dsDNA Assay kit) and 1X TE in 8 tubes as below. The stock tubes can be used multiple times, keep any leftover in the fridge at 4 C between experiments.
Vortex well and spin down the DNA standard before every use. Not vortexing the standards thoroughly will negatively affect the standard curve and concentration readings. Serial dilutions should be prepared as shown in the table below and stored at 4°C :
| A | B | C | D |
|---|---|---|---|
| Tube | Content | Concentration | Volume |
| 1 | 90 μl TE + 10 μl Lambda DNA stock | 10 ng/μl | 100 μl |
| 2 | 50 μl from Tube 1 + 50 μl TE | 5 ng/μl | 100 μl |
| 3 | 50 μl from Tube 2 + 50 μl TE | 2.5 ng/μl | 100 μl |
| 4 | 50 μl from Tube 3 + 50 μl TE | 1.25 ng/μl | 100 μl |
| 5 | 50 μl from Tube 4 + 50 μl TE | 0.625 ng/μl | 100 μl |
| 6 | 50 μl from Tube 5 + 50 μl TE | 0.3125 ng/μl | 100 μl |
| 7 | 50 μl from Tube 6 + 50 μl TE | 0.15625 ng/μl | 100 μl |
| 8 | TE only | blank |
For 384-well plates, prepare a 1:400 solution of PicoGreen™ dsDNA Assay in 1X TE buffer (39µL per sample). Vortex to mix.
Pipette 1µL each of the 7 standards + 1 Blank into a black, flat-bottom 384 MicroWell™ plate. Place the standards on one column.
Pipette 1µL of your samples into the center of each well of the Nunc™ F384 MicroWell™ polystyrene plate.
Add 39µL PicoGreen + TE mix into every well. There is no need to mix.
Cover the plate with the provided plastic (transparent) lid to prevent possible contaminations.
Allow 0h 2m 0s for the dye to bind the DNA. Protect from light but keep at room temperature. For optimal results, the plate should be read within the next hour.
Use a plate reader to measure fluorescence (excitation: 485 nm; emission: 530 nm; read from top; endpoint reading).
Plate normalisation
Prepare a normalisation plate by adding 1µL purified cDNA and nuclease-free water to a final concentration of 200 pg/µL.
Tagmentation and enrichment PCR
This step can be carried out either by using the commercially available Nextera XT kit or an in-house Tn5 transposase , as described below. Indexing primers can be purchased from Illumina
(Nextera XT index kit v2) or ordered from your local oligonucleotide manufacturer.
Indexing primer sequences can found in the "Materials" section.
Tagmentation with in-house Tn5 transposase
| A | B | C | D |
|---|---|---|---|
| Reagent | Volume (μl) | Final Concentration | Volume 384-well plate (μl) |
| Dimethylformamide (DMF) (100%) | 0.8 | 20% | 368.64 |
| TAPS-Mg buffer (5x) pH 7.3 | 0.8 | 10 mM TAPS, 5 mM MgCl2 | 368.64 |
| Tn5 transposase (2 μM working dil.) | 0.1 | 62.5 nmol | 46.08 |
| Nuclease-free water | 1.3 | - | 599.04 |
| Total volume | 3.0 | 1,382.4 |
Dispense 3µL tagmentation mix in a new 384-well plate.
Add 1µL normalised cDNA (200 pg/μl) to each well containing the tagmentation mix.
Seal the plate, vortex, spin down, and carry out the tagmentation reaction: 55°C for
0h 8m 0s , 4°Chold. Upon completion proceed immediately to the next step.
Add 1µL 0.2% SDS to each well. Seal the plate, vortex, spin down and incubate 0h 5m 0s at Room temperature. Do not put the plate back on ice.
Add 2µL N7xx + S5xx index adaptors (5micromolar (µM)) each.
Add 3µL enrichment PCR mix to each well.
| A | B | C | D |
|---|---|---|---|
| Reagent | Per sample (μl) | Final Concentration | Volume 384-well plate (μl) |
| KAPA HiFi enzyme (1 U/µl) | 0.2 | 0.02 U/μl | 92.16 |
| dNTPs (10 mM) | 0.3 | 300 nM | 138.24 |
| KAPA HiFi Buffer (5x) | 2.0 | 1X | 921.60 |
| Nuclease-free water | 0.5 | 230.40 | |
| Total volume | 3.0 | 1,382.40 |
Seal the plate, vortex, spin down (0h 0m 30s, 750x g), and place it in a thermocycler and carry out the enrichment PCR reaction. Adjust the number of PCR cycles according to the number of processed cells.
| A | B | C | D |
|---|---|---|---|
| Step | Temperature | Time | Cycles |
| Gap-filling | 72°C | 3 min | 1 |
| Initial denaturation | 98°C | 30s | 1 |
| Denaturation | 98°C | 10s | 14-16x |
| Annealing | 55°C | 30 sec | |
| Elongation | 72°C | 30 sec | |
| Final Elongation | 72°C | 5 min | 1 |
| Hold | 4°C | Hold |
Library clean-up and quantification
Take an aliquot from each sample well (i.e. 5 µl) and transfer it to a 1.5 mL Eppendorf tube for the final library pool clean-up. The plate containing the rest of the libraries can be stored long-term at -20°C
Remove the Sera-Mag SpeedBeads™ working solution (AMPure XP beads or SPRI beads when using a commercial solution) from the 4°C storage and equilibrate it at room temperature for 0h 15m 0s.
Add 0.8X volume ratio Sera-Mag SpeedBeads™ working solution to the pool. Mix thoroughly by pipetting or vortexing.
Incubate the tube off the magnetic stand for 0h 5m 0s at Room temperature .
Place the tube on the magnetic stand and leave it for 0h 5m 0s or until the solution appears clear.
Remove the supernatant without disturbing the beads.
Recommended: wash the pellet with 1 mL 80% v/v ethanol. Incubate 0h 0m 30s without removing the tube from the magnetic stand.
Remove any trace of ethanol and let the bead pellet dry for 0h 2m 0s or until small cracks appear. Do not cap the tube or remove it from the magnetic stand during this time. Do not completely air-dry the beads.
Remove the tube from the magnetic stand, add 50µL nuclease-free water and mix well by pipetting or vortexing to resuspend the beads.
Incubate0h 2m 0s off the magnetic stand.
Place the tube back on the magnetic stand and incubate for0h 2m 0s or until the solution appears clear.
Remove 49µL of the supernatant and transfer it to a new 1.5-ml LoBind tube. Store the cDNA at -20°C long-term or until ready for sequencing.
Use Qubit fluorometer to quantify the library. Library yield can vary depending on the number of cells being pooled.
Check the final library size on the Agilent Bioanalyzer.
Use the average size indicated on the Bioanalyzer and the concentration reported after Qubit measurement to determine the exact molarity required for sequencing.
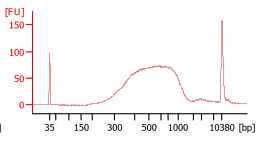
Sequencing
The clean library pool can be sequenced on any Illumina sequencer. Follow the specifications reported for each individual instrument. Single-End 75 bp is generally sufficient but longer read modes or paired-end sequenced are also options depending on the biological question.
Analysis
Data generated with FLASH-seq bulk can be analysed with standard unstranded bulk RNA-sequencing pipelines. We suggest the following tools:
-
(optional) Trim left-over adapters / TSO / oligo-dT with trimmomatic or bbduk.
-
Map reads with STAR.
-
Select properly mapped reads with samtools (-F 260).
-
Visualise the alignment with IGV.
-
Explore the mapping statistics with rseqc (gene body coverage, intron/exon/intergenic mapping).
-
Assign reads to features with featureCounts.
-
Explore isoforms with RSEM or BRIE.
-
Calculate differential expression with DESeq2, EdgeR or limma.
