ATAC Sequencing Protocol
Klaus H. Kaestner Lab, Suzanne Shapira
Abstract
Interrogating cell-type specific chromatin accessibility can reveal cis-regulatory elements linked to downstream gene expression patterns. Assay for Transposase-Accessible Chromatin using Sequencing (ATAC-Seq) is a technique that assays genome-wide chromatin accessibility using a tagmentation protocol that inserts sequencing adapters into open genomic regions. This technique can reveal nucleosome positioning patterns, map enhancer and promoter regions, or reveal transcription factor binding sites. When applied to studies of diabetic patients, this technique can reveal potential differences in genomic accessibility patterns between non-diabetic pancreata compared to diseased organs.
Steps
Resuspension Buffer
1. To use for Lysis Buffer and Wash Buffer, can be stored at room temperature long-term.
500 μl 1M Tris-HCl, pH 7.5 (final 10 mM)100 μl 5 M NaCl (final 10 mM)150 μl 1 M MgCl2 (final 3 mM) 49.25 ml nuclease-free H²O
50mL
Cell Lysis
1. Centrifuge 100,000 cells to pellet [NOTE: The person who is sorting and aliquoting cells will tell you speed/time, to centrifuge usually this is 8,000 xg for 4 minutes] NOTE : The person who is sorting and aliquoting cells will tell you speed/time, to centrifuge usually this is 8,000 xg for 4 minutes], and then discard supernatant.
2. Add 100µL cold Lysis Buffer, pipet up and down 3x gently to resuspend cells. [NOTE: Lysis buffer volume should be scaled to the number of cells; e.g. if you only have 50,000 cells, add NOTE : Lysis buffer volume should be scaled to the number of cells; e.g. if you only have 50,000 cells, add 50µL cold Lysis Buffer]
97 μl Resuspension Buffer1 μl 10% NP-40 (final 0.1% v/v)1 μl 10% Tween-20 (final 0.1% v/v) 1 μl 1% Digitonin (final 0.01% v/v)100µL 3. Incubate on ice x3 minutes. 4. Add 1mL Wash Buffer, invert tube 3 times gently.990 μl Resuspension Buffer 10 μl 10% Tween-20 (final 0.1% v/v)1mL
5. Centrifuge at 500 xg for 10 minutes at 4°C .
6. Discard supernatant (cytoplasm), keep pellet (nuclei).
Transposition
1. While cells are centrifuging, make transposition reaction mix, using the Nextera DNA Library Prep Kit. [NOTE: Transposition reaction mix volume should be scaled to the number of cells; e.g. if you only have 50,000 cells, add NOTE : Transposition reaction mix volume should be scaled to the number of cells; e.g. if you only have 50,000 cells, add 50µL transposition mix]
50 μl 2X TD Buffer (Tagment DNA Buffer)
33 μl 1X PBS
1 μl 10% Tween-20 (final 0.1% v/v)
1 μl 1% Digitonin (final 0.01% v/v)
5 μl Tn5 Transposase (Tagment DNA Enzyme 1)
10 μl nuclease-free H²O
100µL
2. Add transposition reaction mix to pellet, pipet up and down 6x gently to resuspend nuclei.
3 . Incubate at 37°C for 30 minutes on thermomixer at 1,000 rpm.
DNA Purification
1. Isolate DNA using Qiagen MinElute Reaction Cleanup Kit.
2 . Elute DNA in 10 μl EB (Elution Buffer).
Note:
OK to store DNA at -20°C at this point.
PCR Amplification (Library Generation)
1. Combine the following in a PCR tube for each sample:
10 μl purified transposed DNA
10 μl nuclease-free H²O
2.5 μl Ad1_noMX primer (25 μM)
2.5 μl Ad2.* indexing primer (25 μM)
25 μl NEBNext High-Fidelity 2X PCR Master Mix
50µL
2. Amplify samples in PCR machine with following program:
72°C 5 minutes x5 cycles
98°C 30 seconds x5 cycles
98°C 10 seconds x5 cycles
63°C 30 seconds x5 cycles
72°C 1 minute x5 cycles
3. Remove tubes from PCR machine and use 5µL of each partially-amplified library to perform qPCR to determine how many additional PCR cycles are needed. The goal is to stop amplification well prior to saturation to avoid variation among samples due to PCR bias.
5 μl partially-amplified library
3.85 μl nuclease-free H²O
0.5 μl Ad1_noMX primer (25 μM)
0.5 μl Ad2.* indexing primer (25 μM)
0.15 μl 100X SYBR Green I
5 μl NEBNext High-Fidelity 2X PCR Master Mix
15µL
4. Perform qPCR using following program:
98°C 30 seconds x20 cycles
98°C 10 seconds x20 cycles
63°C 30 seconds x20 cycles
72°C 1 minute x20 cycles
5. **qPCR Plate setup:
a. Well type: unknown
b. Collect fluorescence data for ROX and FAM
c. Reference dye: ROX
6. **qPCR Thermal Profile setup:
a. Pre-Melt/RT Segment: 1 plateau
b. Amplification Segment: normal 3 skip
c. Dissociation/Melt segment: uncheck this box
7. Plot R vs Cycle Number. Calculate the number of additional PCR cycles needed for each sample, by determining the number of cycles needed to reach 1/3 of the maximum R.
8 . Continue PCR on remaining 45µL of each partially-amplified library for the appropriate number (N) of cycles:
98°C 30 seconds N cycles
63°C 30 seconds N cycles
72°C 1 minute N cycles
Library Purification
1. Warm AMPure XP beads to room temperature, and vortex for 15 seconds to resuspend.
2. For double-sided bead purification (to remove primer dimers and large >1,000 bp fragments):
a. Transfer each PCR sample to an epi tube, add 0.5X volume (22.5µL ) AMPure XP beads, pipet up and down 10x to mix thoroughly.
b. Incubate at room temperature for 10 minutes.
c. Place epi tubes in magnetic rack for 5 minutes.
d. Transfer supernatant to new epi tube.
e. Add 1.3X original volume (58.5µL ) AMPure XP beads, pipet up and down 10x to mix thoroughly. (This results in a final 1.8X bead buffer:sample ratio.)
f. Incubate at room temperature for 10 minutes.
g. Place epi tubes in magnetic rack for 5 minutes.
h. Discard supernatant.
i. Wash beads with 200µL 80% EtOH (freshly made), pipet EtOH over beads 10x, then discard EtOH.
j. Leave tube on magnetic rack with cap open for 10 minutes.
k. Ensure all EtOH is removed.
l. Resuspend beads in 20µL nuclease-free H²O, pipet up and down 10x to mix thoroughly.
m. Incubate at room temperature for 2 minutes, then quickly spin epi tube down
n. Place epi tube in magnetic rack for 1-5 minutes.
o. Transfer supernatant to new epi tube.
3 . Store purified libraries at -20°C .
Assessing Library Quality
1. Add 1µL of each library to 3µLnuclease-free H²O (to make 1:4 dilution).
2. Run 1µL of each diluted library on an Agilent High Sensitivity DNA Bioanalysis chip.
3. Use 1µL of each diluted library to measure DNA concentration by QuBit.
Sequencing
1. Use 50 bp paired-end (50PE) sequencing.
2. Goal is to obtain >50 million genomic reads per sample minimum to assess open vs closed chromatin regions, and >200 million genomic reads per sample to detect transcription factor binding sites. Remember that many sequencing reads may map to contaminating mitochondrial DNA.
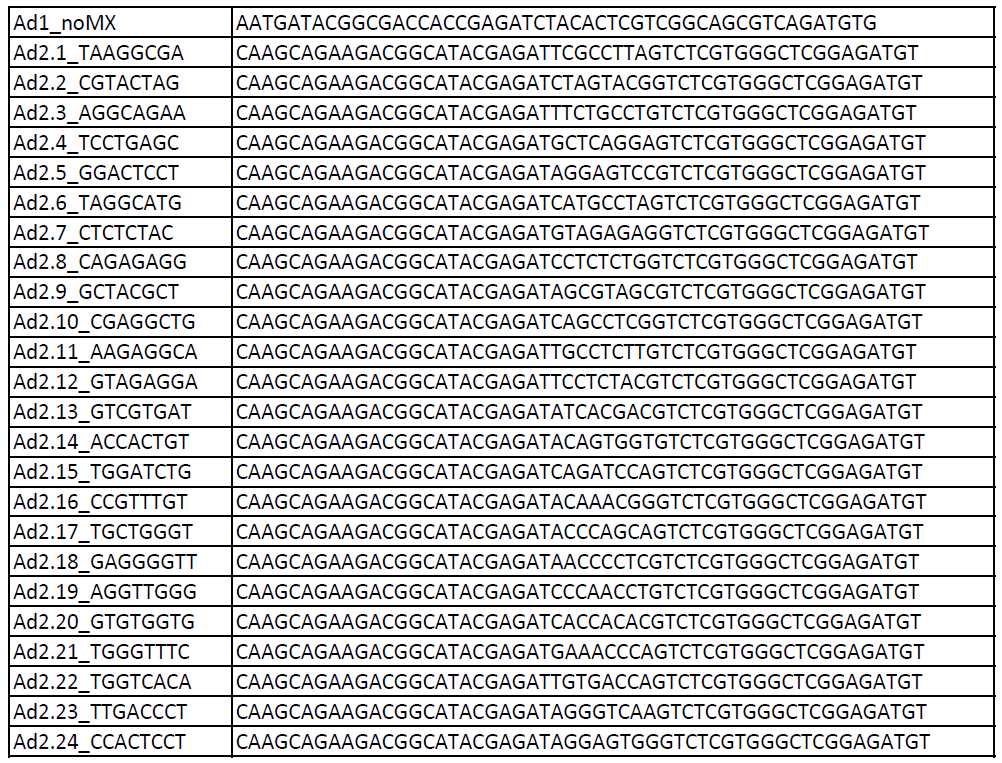
Table of PCR Primers (Illumina/Nextera i5 common adapter and i7 index adapters):