聚合酶链式反应
掌握聚合酶链式反应(PCR)的基本原理。
什么是PCR?
聚合酶链式反应(PCR)是全球实验室中应用最广泛的分子生物学方法之一。通过热循环和耐高温DNA聚合酶的应用,PCR能够对特定的DNA序列进行指数级扩增。
这一反应是由凯利·穆利斯(Kary Mullis)于1983年首次开发此技术,并在1985年与同事首次将这一技术应用于扩增β-球蛋白基因。穆利斯在《酶学方法》出版物中对该方法进行了更为详尽的描述,后来因为其在此方面的贡献,荣获了1993年的诺贝尔化学奖。
PCR所需的5个关键组分如下:
1.DNA模板
2.引物
3.DNA聚合酶
4.dNTPs(脱氧核苷酸三磷酸)
5.反应缓冲液

DNA模板
无论是单链还是双链DNA,都是PCR的标准模板。DNA来源包括基因组DNA(gDNA)、互补DNA(cDNA)或质粒。
引物
PCR引物通常设计一对上下游引物,分别称为正向引物和反向引物。这些引物与单链DNA的互补区域相结合,一旦结合到靶标区域,PCR引物就可在DNA聚合酶作用下延伸新序列。
DNA聚合酶
PCR中常用的聚合酶类型被称为Taq DNA聚合酶。该聚合酶得名于其最初提取的嗜热菌——水生栖热菌(Thermus aquaticus),Taq聚合酶能在高温下高效工作。
您可以在下方了解更多关于DNA聚合酶的特性。
dNTPs(核苷酸)
为了在PCR过程中生成新的DNA链,必须具备四种脱氧核苷三磷酸(dNTP)碱基——腺嘌呤(ATP)、胸腺嘧啶(TTP)、胞嘧啶(CTP)和鸟嘌呤(GTP)。
反应缓冲液
PCR反应缓冲液包含了成功进行反应所需的基本成分,其中包括DNA聚合酶辅助因子镁离子(Mg2+),有助于聚合期间dNTP的结合,可以为聚合酶提供最佳工作条件。
PCR的工作原理是什么?
一个典型的PCR过程包含以下步骤:
第一步:预变性
第二步:变性
第三步:退火
第四步:延伸
变性、退火及延伸(步骤2至4)构成了PCR循环,这一循环会重复多次以增加最终PCR产物的数量。
步骤1:预变性
双链DNA首先被加热至约94°C,持续几分钟。高温破坏了双链互补碱基之间的氢键,从而形成两条单链DNA。
DNA模板越大,预变性阶段所需的时间就越长。例如,相比于cDNA,基因组DNA(gDNA)含有更多的碱基和氢键,因此需要适当延长gDNA预变性时间,确保双链DNA完全解离。
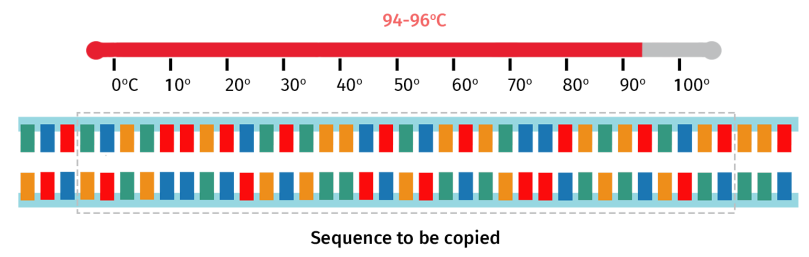
(要复制的序列)
步骤2:变性
在每次PCR循环开始时,样品会被加热至94°C左右,这个过程只有几秒钟。这个步骤能够确保新形成的双链PCR产物重新变性为单链形式。
步骤3:退火
随着目标模板变为单链形式,样品随后温度降至45–60°C,从而使PCR引物与其在模板上的互补区域退火结合。
确切的退火温度因引物长度和GC含量等性质而变化。例如,长度较长的PCR引物通常需要较高的退火温度,而较短长度的引物所需的退火温度则较低。
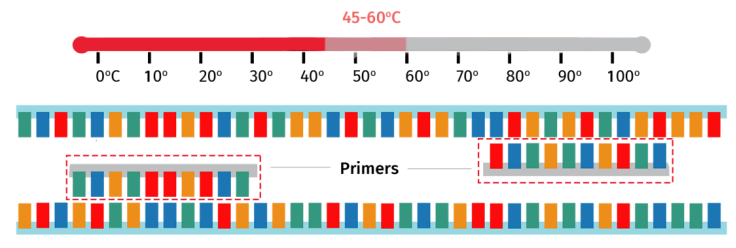
步骤4:延伸
接下来将样品加热至72°C,这个温度是Taq聚合酶能够从已结合引物的3末端开始延申合成新的DNA链的最佳温度。
在延伸步骤结束时,PCR产物的拷贝数相比于PCR循环开始时会翻倍。

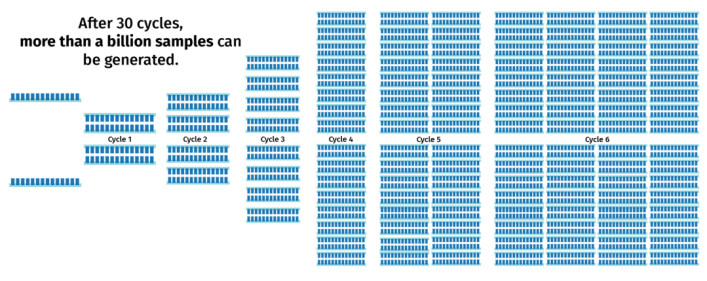
PCR延伸
随后PCR循环(步骤2至4)会重复进行,通常循环30至40次,增加PCR片段的拷贝数量。在每一次循环后,产物拷贝的数量都会翻倍。这意味着经过30次循环的PCR反应可以从一条模板扩增得到超过10亿倍的产物!
DNA聚合酶:四大关键属性
目前市面上有多种不同类型的DNA聚合酶可供选择,每种都有自己独特的性质。因此,在考虑PCR应用时,需关注聚合酶的四个关键属性:
1.热稳定性
2.特异性
3.延申合成能力
4.保真度:衡量聚合酶在复制DNA过程中引入错误的频率,即复制准确性。
在某些情况下,为了优化热稳定性、特异性、延申性和保真度的要求,可能更适合使用不同聚合酶的混合物。
热稳定性
由于热循环是DNA扩增重复链式反应条件的关键特征,因此,所使用DNA聚合酶的热稳定性非常重要。尽管最初来自于嗜热菌菌株的 Taq DNA 聚合酶可耐受相对较高的温度,但是其半衰期在90°C以上时明显缩短。当使用 长时间高温 使具有二级结构和富含GC序列的DNA变性时,这一缺陷便成为一大难题。同样,在扩增长片段模板时,需要更大量的Taq DNA 聚合酶或补充Taq DNA聚合酶,以供长时间孵育。因此,从超嗜热菌分离的DNA聚合酶具有更高的热稳定性,将有助于克服这些挑战。
Pfu DNA聚合酶是一种为大家所熟知的超耐热酶,来自于水热环境中的超嗜热古细菌 Pyrococcus furiosus 。在95°C下,Pfu聚合酶的稳定性能比 Taq 聚合酶高20倍。其它常见的超耐热DNA聚合酶包括来自古细菌 Thermococcus 和 Pyrococcus 的KOD和GBD,其中GB-D的超嗜热DNA聚合酶Deep Vent,在98–100°C下的半衰期可达数小时。
尽管古细菌DNA聚合酶的热稳定性极强,但它们在某些方面也具有一定局限性。例如,超耐热 Pfu DNA 聚合酶的合成能力较低(与 Taq DNA聚合酶相比),因此合成DNA的速度较慢,这就需要延长延申时间。此外,古细菌DNA聚合酶无法扩增含有尿嘧啶的DNA模板,因为其存在尿嘧啶结合区域作为一种DNA修复机制。含尿嘧啶的DNA序列是 防止PCR 残余污染 和经亚硫酸氢盐转化进行 基因座甲基化分析 的基础。
特异性
非特异性扩增是PCR反应最主要的障碍之一,因为它会显著降低目的基因扩增的产率和灵敏度,从而影响扩增结果的合理解释和下游实验应用的成功。而DNA聚合酶经常会延伸错配的目的基因和引物二聚体,这是非特异性扩增的常见来源。如何减少非特异性扩增?第一张方案是在冰上配制PCR反应,这样有助于将DNA聚合酶的活性保持在低水平,但在PCR开始之前依然可能会合成不需要的产物。
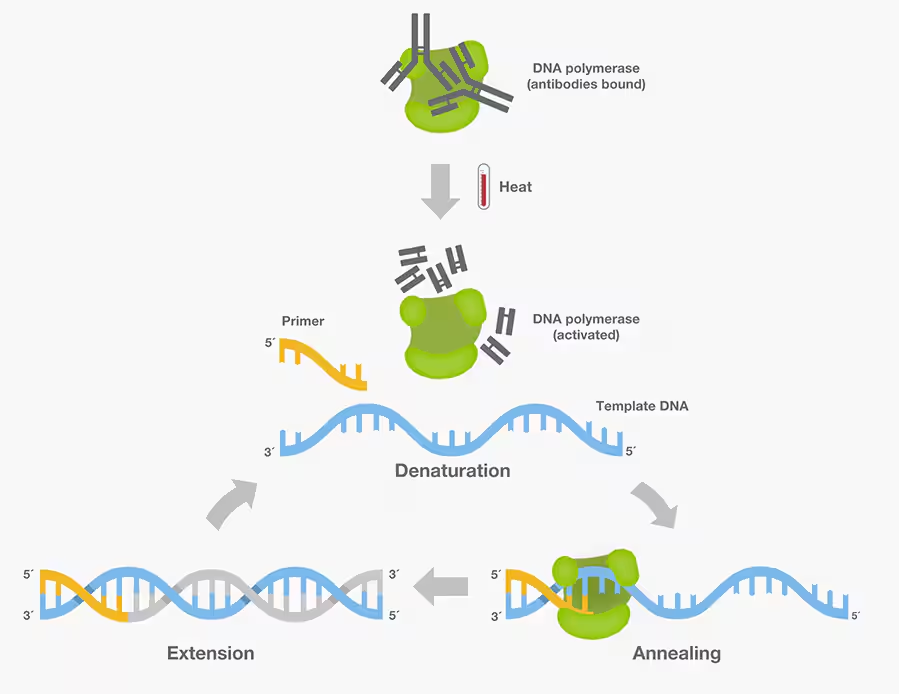
提高DNA聚合酶和PCR特异性的另一种方案是使用热启动Taq。热启动Taq混合物通常包含一种与抗体结合的DNA聚合酶,这种抗体在低于90°C的温度下抑制Taq的活性。在PCR的初始变性步骤中,高温会使抗体从聚合酶上解离,从而激活Taq酶。接下来,样品将被加热至72°C,这个温度提供了最佳的延伸条件,使Taq聚合酶能够从已结合引物的3末端开始合成新的DNA链。因为只有在90℃以上的预变性步骤之后才能开始扩增,所以该技术被称为“热启动”。
聚合酶延伸合成能力
聚合酶的延伸性是指单一酶在从DNA模板上脱落之前能连续掺入的核苷酸数目。
在75°C条件下,天然Taq聚合酶通常能够以每秒10到45个核苷酸的速度扩增DNA,也就是说大约每分钟扩增2千个碱基!
有的DNA聚合酶经过工程化改造改善了其DNA结合结构域,使其比传统的Taq聚合酶更加稳定。例如,KAPA2G聚合酶的速度约为每秒掺入150个核苷酸,比Taq聚合酶高出3倍。
提高聚合酶延伸性能对于扩增长片段PCR产物(例如大于10千碱基)是有益的,还有助于推动反应向富含GC碱基对的DNA区域或具有高度二级结构的区域延申。
保真度
保真度指的是DNA聚合酶准确复制目标模板而不会掺入错误碱基的能力。
先前对Taq聚合酶的分析表明,其总体错误率为1.8 x 10^-4,这意味着由Taq聚合酶掺入的每10,000个碱基中,至少会出现1个错误(包括碱基替换、缺失、插入等)。Taq聚合酶的保真度相对较低与其缺乏3'至5'外切酶校读功能有关。
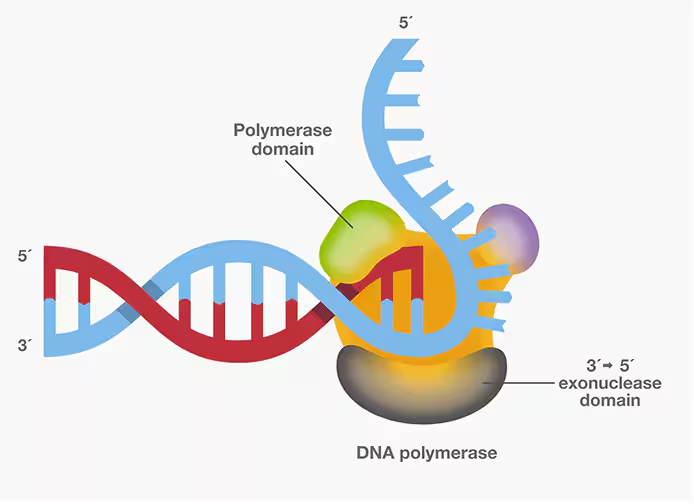
高保真DNA聚合酶,具备基于 3′ → 5′核酸外切酶校正活性,可校正错误插入的核苷酸。DNA聚合酶上的核酸外切酶活性位点与其5′→ 3′聚合酶活性位点是分离的。当错配的核苷酸插入聚合结构域,DNA合成将因不合适的碱基配对动力学而暂停。暂停期间,DNA聚合酶将切除错配的核苷酸并用正确的核苷酸替换。
对于需要精确克隆长序列的PCR应用,建议使用高保真度聚合酶。像Pfu和Q5®这类聚合酶,具备校读活性,其保真度比Taq聚合酶高出1至2个数量级。
聚合酶链式反应(PCR)的应用
PCR是一种多用途的技术,已经在许多分子生物学应用中得到利用,包括:
1.克隆
2.突变体产生
3.基因型分析
4.基因表达分析
克隆
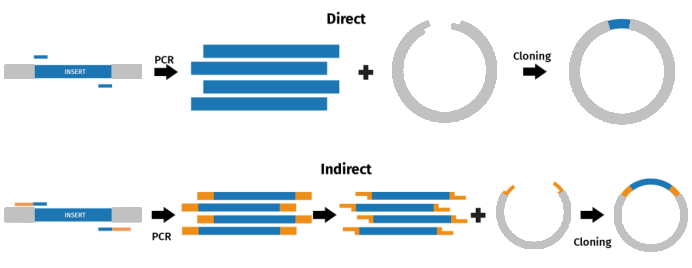
多种克隆方法都在采用PCR技术来直接或间接地重组DNA片段。
直接PCR克隆方法包括TA克隆、GC克隆以及TOPO®克隆。这些方法能够直接克隆PCR产物。其中,TA克隆法利用了Taq聚合酶介导的PCR反应完成后,在产物末端形成的3’A突出,这种黏末端能够与含有3’端T突出的DNA片段(如线性化载体)进行重组。
在间接PCR克隆中,PCR产物在与其他DNA序列重组之前需要进行修饰。一种常见的修饰方法是在PCR引物设计时,引入限制性酶切位点,通过对PCR产物和目标载体进行酶切,从而实现与载体的重组。通过这种间接PCR克隆的方法,可以实现对特定DNA序列的快速、高效的克隆。
突变体产生
PCR突变体产生是一种用于生成定点突变的技术,例如碱基置换、插入和删除等。
举个例子,如果想要通过突变来产生单个碱基的变异,通常会设计一对包含所需突变碱基的PCR引物。突变位点一般位于引物序列的中间。然后使用这对突变引物和高度保真度的DNA聚合酶进行PCR反应,在反应过程中突变位点会被插入到目标序列中,最终形成所需的突变序列。

在衍因智研云软件中,您可以设计诱变引物并模拟突变体产生反应。
基因型鉴定
等位基因特异性PCR是一种用于检测序列变异的方法,可以确定生物体的基因型。
对于等位基因特异性PCR,引物设计用于侧翼目标区域。例如,在创建基因敲除小鼠时,引物用于靶向野生型和转基因基因区域。然后,您可以通过分析琼脂糖凝胶上的PCR产物来确定敲除实验是否成功。
使用衍因智研云,您可以模拟琼脂糖凝胶电泳,预测PCR及限制性酶切后产物带的数量和大小。
基因表达分析
聚合酶链反应(PCR)最普遍的应用之一是通过定量PCR(qPCR)进行基因表达分析,也称为实时PCR(RT-PCR)。
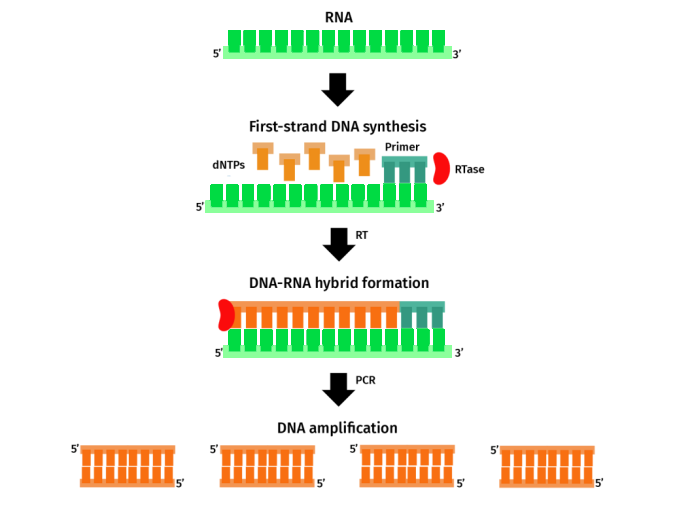
在两步法qPCR中,首先将含有信使RNA(mRNA)转录本的RNA通过反转录转化为互补DNA(cDNA),随后,得到的cDNA作为模板用于qPCR。PCR引物被设计用于扩增感兴趣基因的转录本片段。在qPCR过程中,通过对荧光信号的实时监测来检测转录本的扩增情况,该荧光信号进而被用来确定目标基因的初始表达水平。
在两步法qPCR中,首先将含有mRNA转录本的RNA进行反转录,将其转化为cDNA。随后,以得到的cDNA作为qPCR的模板,并设计用于扩增目的基因转录本的PCR引物,进行扩增。在qPCR过程中,通过实时监测染料法或探针法的荧光信号,来检测转录本的扩增情况,并据此确定目标基因的初始表达水平。
使用衍因智研云,您可以设计引物并模拟PCR实验来规划qPCR实验。
PCR类型
有一些技术是对传统PCR方法的改变,包括重叠延伸PCR和反向PCR。
重叠延伸PCR
重叠延伸PCR,也称为通过重叠延伸剪接,是一种多用途的技术,通常用于在无缝反应中将多个DNA片段融合在一起。
该反应分为三个独立的PCR步骤:延伸、重叠和纯化。
延伸PCR:引物对在相邻DNA片段间引入重叠末端
重叠PCR:延伸PCR的产物随后用于另一次PCR中,以连接重叠的PCR产物
纯化PCR:结合至首个和最后一个DNA片段末端的引物用于新的PCR中,以扩增融合产物
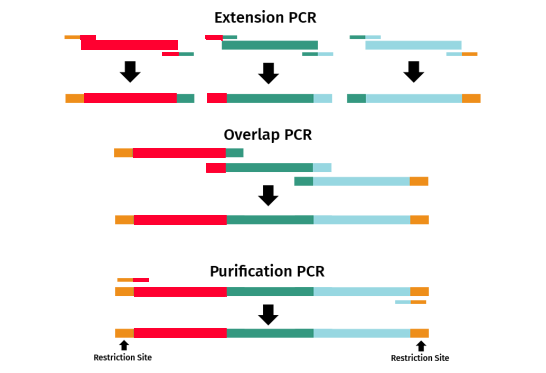
通过使用诱变引物,重叠延伸PCR也可用于引入定点突变。
衍因智研云能协助您设计用于重叠延伸PCR的引物并模拟该反应。
反向PCR
反向PCR用于通过使用与常规PCR引物反向相反的引物,扩增与已知序列相邻的未知DNA序列区域。
反向PCR的一个应用实例是克隆未知序列。一段DNA区域,其中包含一段已知序列,已知序列两侧为未知序列。扩增前先用限制性内切酶酶切样品DNA,然后用DNA连接酶连接成一个环状DNA分子。环化后,利用已知序列区域的引物进行反向PCR,得到的PCR产物可随后连接到克隆载体上,进行分离及测序。
PCR实验成功技巧
防止污染
PCR是一项非常敏感的技术,容易受到诸如gDNA等非目标核酸的污染。
以下是一些简单的方法,可帮助您减少PCR污染的可能性。
1.在每次PCR实验之前,使用脱氧核糖核酸清除剂(如DNAZap™)清洁移液器和工作环境。
2.准备PCR混合液时使用PCR级水。
3.使用带有防气溶胶滤器的吸头。
4.将工作区域分开为不同的区域;在一个房间配置PCR混合液,在另一个房间吸取模板。
5.将PCR试剂和PCR引物分装成工作体积。
6.避免将PCR试剂和移液器用于非PCR相关的实验。
配置PCR主混合液
在涉及多孔反应的PCR实验中,一个非常有用的策略是准备包含DNA聚合酶、反应缓冲液(如dNTPs、盐类等)和引物的主混合液。主混合液可以在一个单独的管中配制,然后等体积分配到每个反应孔中。接下来只需向每个孔中加入所需的模板即可。通过采用这种方式,可以提高实验的效率并减少潜在的误差来源。
主混合液确保了PCR各组分在不同孔之间均匀分布。相比于将每种组分单独移液到每个孔,使用主混合液还可以节省时间。
为了应对在准备主混合液时可能出现的移液误差,建议制备比实际需求多10%的混合液。例如,如果需要准备100个孔,建议制备足够110个孔的主混合液。这样可以确保有足够的混合液可以使用,避免因移液误差导致不够的情况发生。
使用梯度PCR确认最佳退火温度
在使用衍因智研云等计算工具设计PCR引物时,可以计算出估计的引物熔解温度(Tm)。进而,可以根据引物的熔解温度估算PCR的退火温度。
预估的退火温度会因为所用的DNA聚合酶而有所不同:
多数DNA聚合酶:退火温度一般比引物熔解温度低0-5°C
具有校正功能的DNA聚合酶(例如Phusion®、Phire®、Q5®):退火温度比引物熔解温度高6-12°C
当然,这是理论上的PCR退火温度。还有其他因素可能影响反应的退火温度,比如引物浓度和反应缓冲液的组成。因此,对于新设计的引物对,在优化退火温度时建议使用梯度PCR。
梯度PCR是指在相同的PCR循环条件下,通过在不同样本孔之间改变退火温度来实现不同反应条件的PCR。大多数热循环仪都配备了梯度功能,可以在不同样本孔之间设置不同的退火温度。在进行梯度PCR时,建议设置的温度区间应该包含预估退火温度上下5°C的范围。这样可以确保在不同样本孔中都能够寻找到最适合的退火温度条件。
确保DNA模板溶液中不含PCR抑制剂
在提取和准备用于PCR的模板DNA时,要确保其不含可能抑制PCR反应的化合物。
模板溶液中常见的PCR抑制剂包括:
1.蛋白质
2.乙二胺四乙酸(EDTA)
3.乙醇
4.离子型去垢剂(如十二烷基硫酸钠[SDS])
避免添加过多模板
向PCR中添加过多的DNA模板实际上可能会抑制反应。因此,应根据DNA类型调整模板的添加量。
以下可作为决定PCR中DNA模板添加量的参考:
cDNA:1–100 ng
基因组DNA:5–50 ng
质粒DNA:0.1–10 ng
请切记,PCR是一种非常灵敏的技术,因此通常来说,添加比预想中更低浓度的DNA模板会更安全,而不是过量添加。